[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/CLUSTERdeconRes"Penalised regression improves imputation of cell-type specific expression using RNA-seq data from mixed cell populations compared to domain-specific methods
1 Main Figures
1.1 Figure1, multi-response LASSO/ridge
- multi-response LASSO/ridge to predict sample-level cell-type expression
1.2 Figure 2, Data and study design
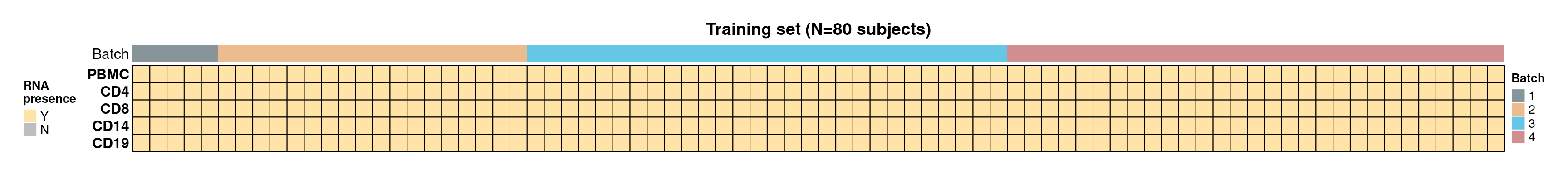
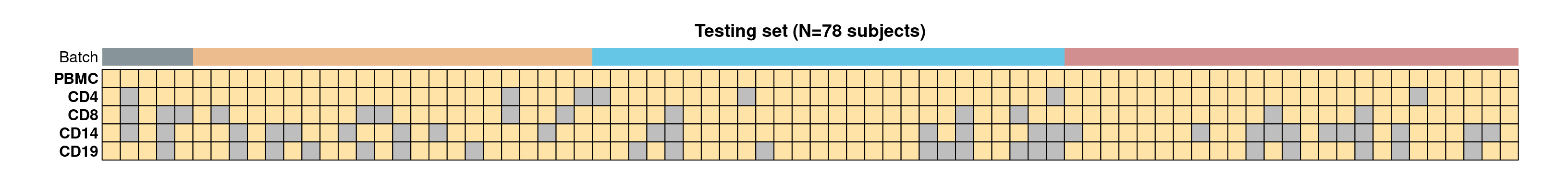
- CLUSTER samples by cell type (row) and subject (column). Cells are coloured based on the availability of RNA (Y for yes, N for no), and the top panel annotations indicate the RNA sequencing batch (Batch).
- Data analysis workflow. Transcripts per million (TPM) were calculated after excluding low-expressed genes. TPM from sorted cells (CD4, CD8, CD14, and CD19) from 80 training samples were used to generate custom signature genes using the CIBERSORTxFractions module. We deconvoluted the cell fractions from PBMC based on inbuilt and custom signatures using CIBERSORTx, using the custom signature genes with bMIND and cell-type specific genes using debCAM. Estimates of cell fractions were compared to the ground-truth cell fractions from flow cytometry, and we assessed fraction accuracy using Pearson correlation and RMSE (root mean square error). Next, we estimated sample-level cell-type gene expression based on inbuilt and custom signature matrices using the CIBERSORTx high resolution module. In parallel, we ran bMIND and swCAM, with the flow cytometry cell fractions, in a supervised mode for estimating cell-type expression. For each cell type, we trained a LASSO/RIDGE model on PBMC and sorted cells with 5-fold cross-validation and used this to predict cell-type gene expression in the test samples. We compared imputed cell-type expressions with the observed ones and evaluated and benchmarked the performance using Pearson correlation, RMSE and a novel measure, differential gene expression (DGE) recovery.
1.3 Figure 3, Prediction accuracy of cell fractions
Code
####################
## flow fractions ##
####################
fractflow <- file.path(
data_dir,
"CLUSTERData/flowFract_wide.rds"
) %>%
readRDS()
## cibx-LM22
#############################
## fractions based on LM22 ##
#############################
lm22merged <- "data4repo/mergedClasses.csv" %>%
file.path(here::here(), .) %>%
scan(., what = "character", sep = "\t")
res_d <- file.path("CIBXres/LM22deconv", runid) %>%
file.path(data_dir, .)
res_d
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/CLUSTERdeconRes/CIBXres/LM22deconv/cluster"
lm22mtx <- file.path(res_d, "sigMTX4dev.txt") %>%
fread()
lm22res <- file.path(res_d, "CIBERSORTxGEP_NA_Fractions-Adjusted.txt") %>%
fread()
stopifnot(names(lm22res)[-1][1:length(names(lm22mtx)[-1])] == names(lm22mtx)[-1])
## fractions in 22 cell types
lm22res.mtx <- lm22res[, names(lm22mtx)[-1], with = FALSE] %>% as.matrix()
rownames(lm22res.mtx) <- lm22res$Mixture
## fractions merged into 4 cell types based on lm22merged
lm22res.4ct <- matrix(NA, nrow = nrow(lm22res.mtx), ncol = length(sctorder))
rownames(lm22res.4ct) <- lm22res$Mixture
for (n in seq_along(sctorder)) {
colidx <- which(lm22merged == sctorder[n])
lm22res.4ct[, n] <- rowSums(lm22res.mtx[, colidx, drop = FALSE])
}
## merged class for 4 sorted cells
ctlm22pooled <- lapply(sctorder, function(ct) {
colnames(lm22res.mtx)[lm22merged == ct]
})
names(ctlm22pooled) <- sctorder
## fractions of 4 cell types scaled to 1
lm22res.4ct.scaled <- apply(lm22res.4ct, 1, function(x) 1 / sum(x) * x) %>% t()
colnames(lm22res.4ct.scaled) <- sctorder
lm22res.4ct.scaled <- lm22res.4ct.scaled[rownames(fractflow), ]
##################
## sorted cells ##
##################
res_d <- file.path("CIBXres/deconv", runid) %>%
file.path(data_dir, .)
res_d
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/CLUSTERdeconRes/CIBXres/deconv/cluster"
customfract <- file.path(res_d, "CIBERSORTxGEP_NA_Fractions.txt") %>%
fread()
customres <- customfract[, ..sctorder] %>% as.matrix()
rownames(customres) <- customfract$Mixture
customres <- customres[rownames(fractflow), ]
###########
## bMIND ##
###########
res_d <- file.path("bMINDres", runid) %>%
file.path(data_dir, .)
res_d
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/CLUSTERdeconRes/bMINDres/cluster"
bmindresFract <- readRDS(file.path(res_d, "bMIND_Fractres.rds"))[["frac"]]
bmindresFract <- bmindresFract[rownames(fractflow), ]
############
## debCAM ##
############
res_d <- file.path("swCAMres", runid) %>%
file.path(data_dir, .)
res_d
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/CLUSTERdeconRes/swCAMres/cluster"
## 4 fold changes (FC) for marker selection
debCAMresFract <- readRDS(file.path(res_d, "debCAM_FractresPure80.rds"))
debCAMresFract %>% names() # OVER.FC_1, FC_2, FC_5, FC_10
[1] "OVE.FC_1" "OVE.FC_2" "OVE.FC_5" "OVE.FC_10"
## go for fraction results based on FC10
debFract <- debCAMresFract$OVE.FC_10$Aest[rownames(fractflow), ]
#######################
## flow + pred fract ##
#######################
## true fraction longformat
flowfract.m <- copy(fractflow) %>%
setDT(., keep.rownames = TRUE) %>%
melt(
data = .,
id.vars = c("rn"),
measure.vars = sctorder,
variable.name = "celltype",
variable.factor = FALSE,
value.name = "flowfract"
) %>%
setnames(., "rn", "sampleName")
## pred fraction in
fract_appro <- c("CIBX-inbuilt", "CIBX-custom", "bMIND-custom", "debCAM")
predfract <- list(
lm22res.4ct.scaled,
customres,
bmindresFract,
debFract
)
names(predfract) <- fract_appro
pltfract <- lapply(names(predfract), function(dat) {
dsn <- as.data.frame(predfract[[dat]]) %>% setDT(., keep.rownames = TRUE)
dsn.m <- melt(
data = dsn,
id.vars = c("rn"),
measure.vars = sctorder,
variable.name = "celltype",
variable.factor = FALSE,
value.name = "predfract",
)
dsn.m[, frt_type := dat]
setnames(dsn.m, "rn", "sampleName")
dsn.m[flowfract.m, flowfract := i.flowfract, on = c("sampleName", "celltype")]
dsn.m
}) %>% rbindlist(., use.names = TRUE)
## limited to test samples
pltfract_test <- pltfract[sampleName %in% sampdata[samtype != "refsamp", sample], ]
pltfract_test[, ":="(
frt_type = factor(frt_type, levels = fract_appro),
celltype = factor(celltype, levels = sctorder)
)]
## r & rmse
cordsn <- pltfract_test[,
{
rhores <- cor(flowfract, predfract)
rmse <- sqrt(mean((flowfract - predfract)^2))
list(rhores, rmse)
},
by = c("frt_type", "celltype")
][, tlab := paste0("R=", round(rhores, 2), "\n", "RMSE=", round(rmse, 3))]
fig2plt <- ggpubr::ggscatter(
data = pltfract_test,
x = "flowfract", y = "predfract",
fill = "celltype", shape = 21, size = 2,
facet.by = c("frt_type", "celltype"),
alpha = 1,
palette = ctfrat.pal,
position = "jitter",
xlab = "Flow Fraction",
ylab = "Predicted Fraction"
) +
geom_abline(slope = 1, intercept = 0, linetype = 2, color = "blue") +
facet_grid(frt_type ~ celltype, scales = "fixed") +
ggpp::geom_text_npc(
aes(
npcx = 0.05, npcy = 0.97,
label = tlab
),
size = 3.5,
data = cordsn
) +
used_ggthemes(
strip.text.y.right = element_text(angle = 0),
legend.position = "none"
)- Prediction accuracy of cell fractions by cell type (column) and approaches (row). Pearson correlation (R) and root mean square errors (RMSE) were calculated between estimated fractions (y-axis) and flow cytometry measures (x-axis). Each point is a testing sample and dashed blue lines indicate \(y = x\). CIBX-inbuilt: CIBERSORTx fraction deconvolution using the inbuilt signature matrix; CIBX-custom: CIBERSORTx fraction deconvolution using the custom signature matrix; bMIND-custom: bMIND fraction estimation using the custom signature matrix; debCAM-custom: debCAM fraction estimation using cell-type specific genes.
Code
fig2plt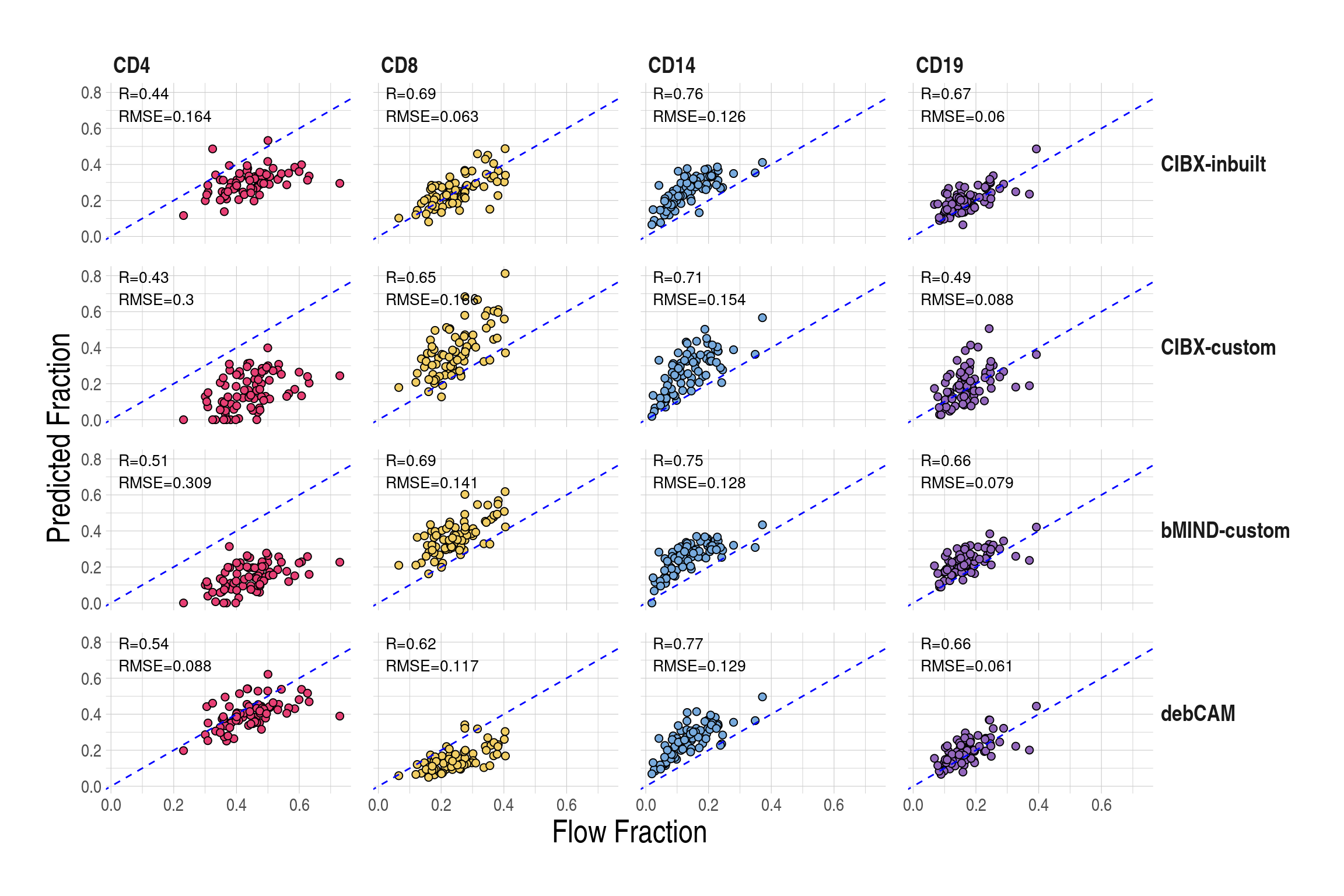
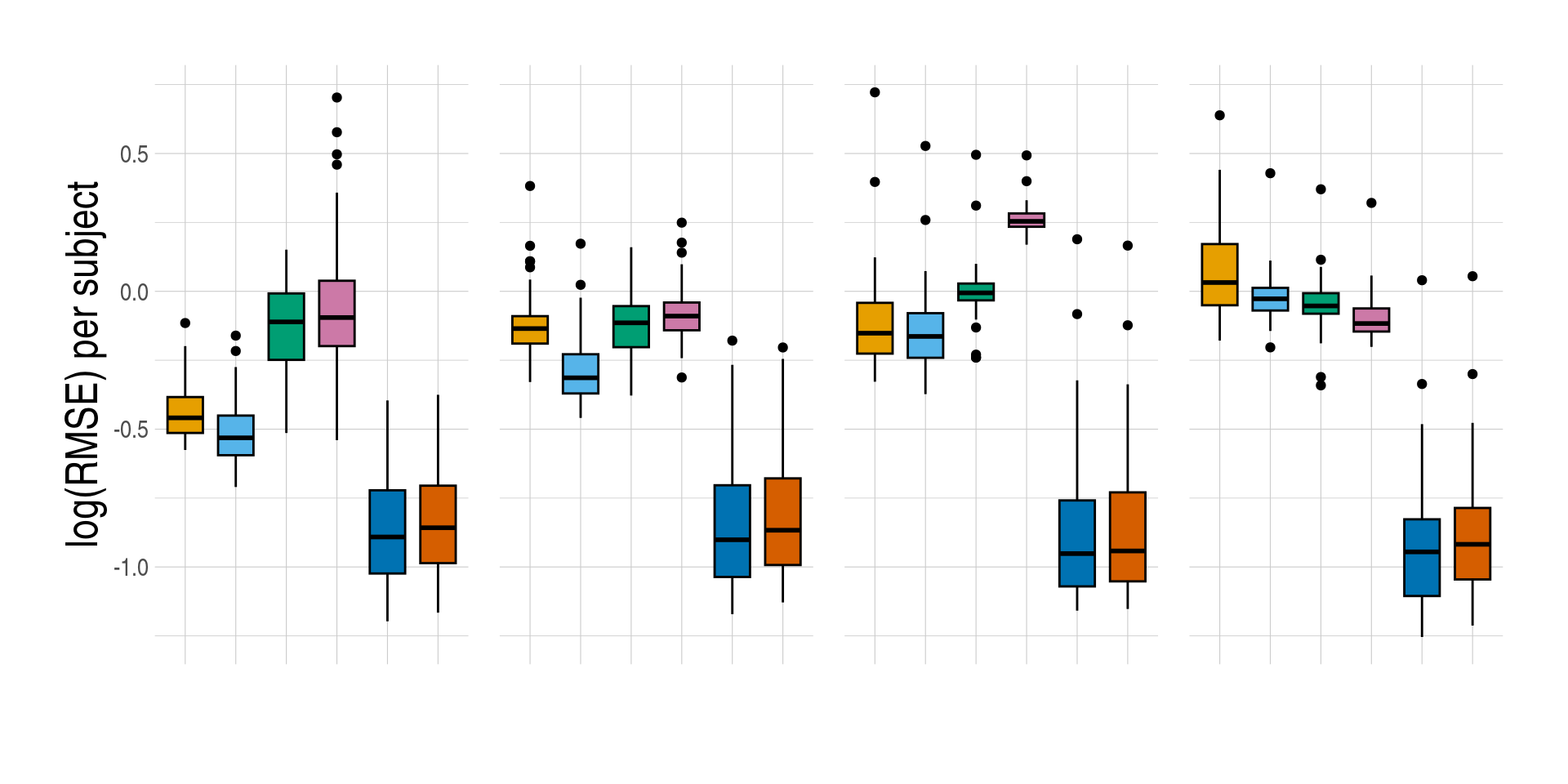
1.4 Figure 4, Prediction accuracy of cell-type expression
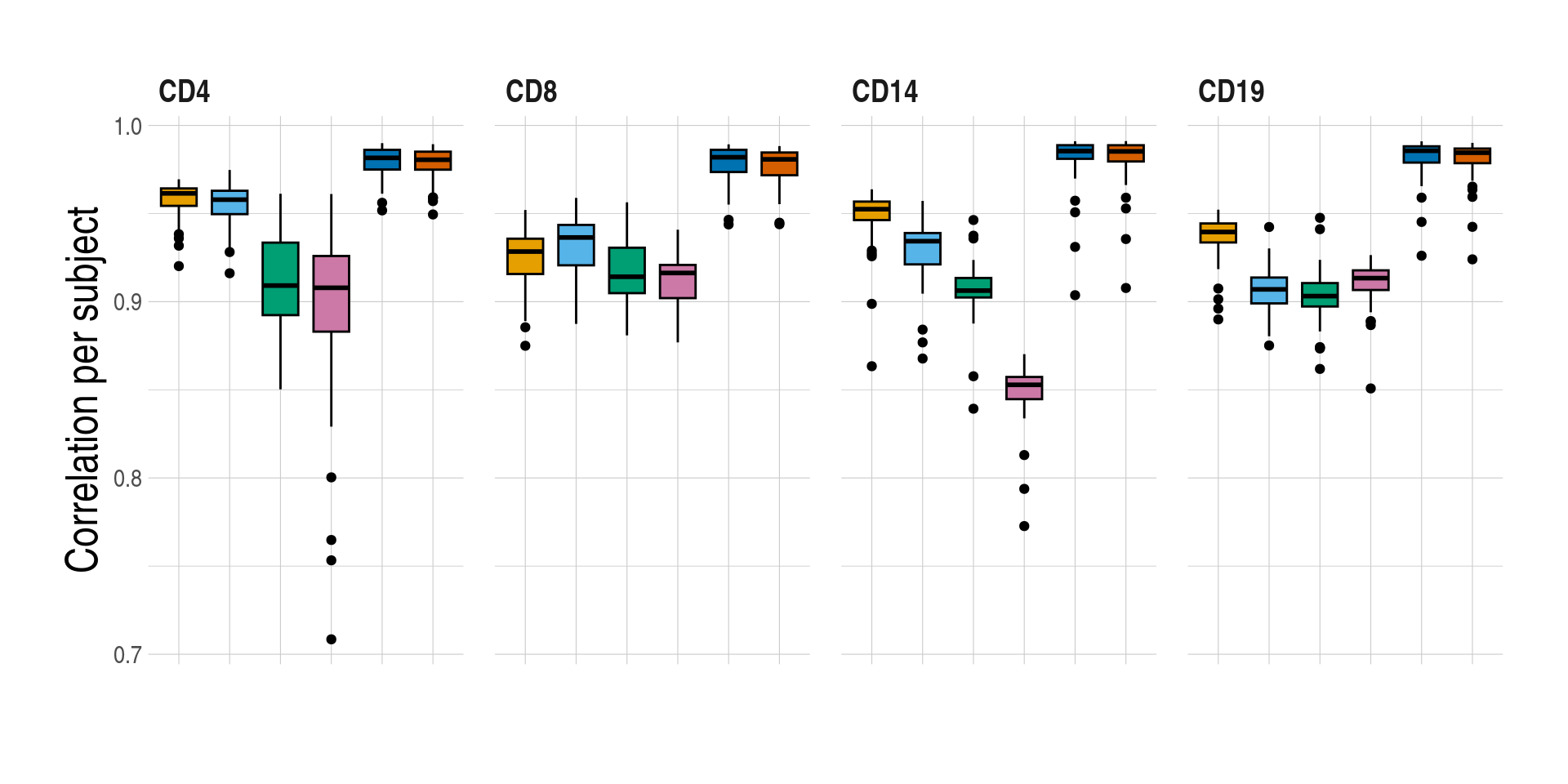
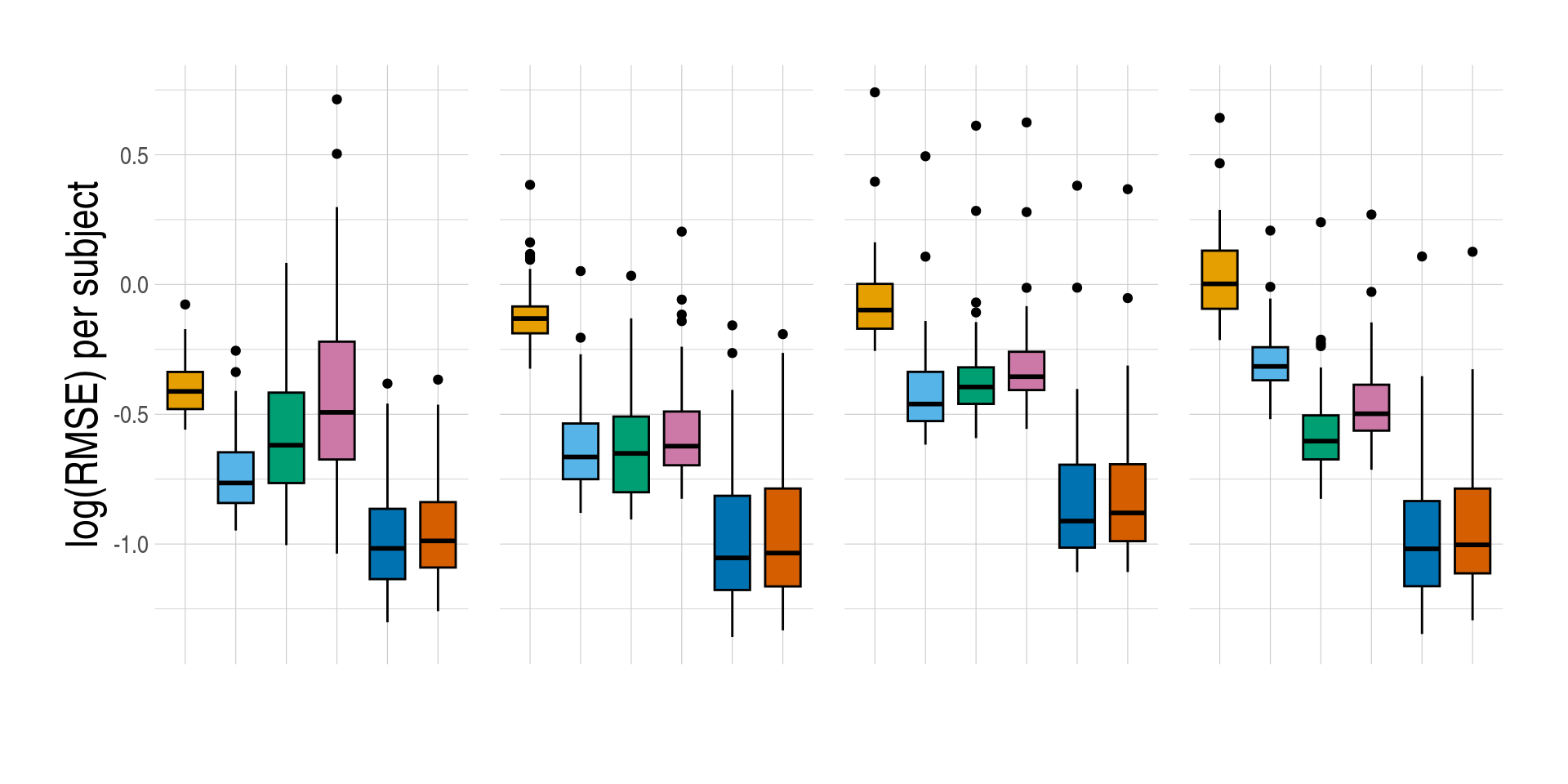
- Distributions of prediction accuracy by cell type and approach. Prediction accuracy was evaluated by calculating Pearson correlation and root mean square error (RMSE) between observed and predicted cell-type expression across genes for the same subjects by cell type and approach. The approaches used were: inbuilt - CIBERSORTx expression deconvolution with the inbuilt signature matrix, custom - CIBERSORTx expression deconvolution with a custom signature matrix derived from sorted cell-type expression in training samples, bMIND - bMIND expression deconvolution with flow fractions, swCAM - swCAM deconvolution with flow fractions, and LASSO/RIDGE - expression predicted from regularised multi-response Gaussian models.
Code
## expression from approaches
cdata_explst <- prep_dat(
rid = "cluster",
res_dir = data_dir,
cluster = TRUE
)
## predicted accuracy
numCores <- length(expr_order)
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
pedaccdsn_persub <- foreach(i = expr_order, .combine = "rbind") %dopar% {
run_fig3v(
obvexpr = cdata_explst$oexpr, impvexpr = cdata_explst$iexpr,
sce = i, ct = sctorder, persub = TRUE
) %>%
rbindlist()
}
stopCluster(cl)
pedaccdsn_persub[, .N, by = c("approach", "celltype")]
pedaccdsn_persub[, ":="(
approach = factor(approach, levels = expr_order),
celltype = factor(celltype, levels = sctorder)
)][, measure_rmse := log(measure_rmse)]
measureii <- c("Correlation per subject", "log(RMSE) per subject")
names(measureii) <- c("measure_cor", "measure_rmse")
lapply(seq_along(measureii), function(idx) {
bplt <- ggboxplot(
data = pedaccdsn_persub,
x = "approach",
xlab = "",
y = names(measureii)[idx],
ylab = measureii[idx],
fill = "approach",
palette = appro.cols %>% as.vector(),
facet.by = c("celltype")
) +
facet_grid(~celltype, scales = "free")
if (idx == 1) {
bplt <- bplt +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "none"
)
} else {
bplt <- bplt +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "none",
strip.text = element_blank()
)
}
bplt
})
Code
numCores <- length(expr_order)
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
pedaccdsn <- foreach(i = expr_order, .combine = "rbind") %dopar% {
run_fig3v(
obvexpr = cdata_explst$oexpr, impvexpr = cdata_explst$iexpr,
sce = i, ct = sctorder, persub = FALSE
) %>%
rbindlist()
}
stopCluster(cl)
pedaccdsn[, .N, by = c("approach", "celltype")]
approach celltype N
1: inbuilt CD4 6261
2: inbuilt CD8 8003
3: inbuilt CD14 7522
4: inbuilt CD19 5779
5: custom CD4 7241
6: custom CD8 11794
7: custom CD14 7823
8: custom CD19 5881
9: bMIND CD4 18871
10: bMIND CD8 18871
11: bMIND CD14 18821
12: bMIND CD19 18864
13: swCAM CD4 18871
14: swCAM CD8 18870
15: swCAM CD14 18867
16: swCAM CD19 18871
17: LASSO CD4 18871
18: LASSO CD8 18871
19: LASSO CD14 18053
20: LASSO CD19 18854
21: RIDGE CD4 18871
22: RIDGE CD8 18871
23: RIDGE CD14 18555
24: RIDGE CD19 18747
approach celltype N
pedaccdsn[, ":="(
approach = factor(approach, levels = expr_order),
celltype = factor(celltype, levels = sctorder)
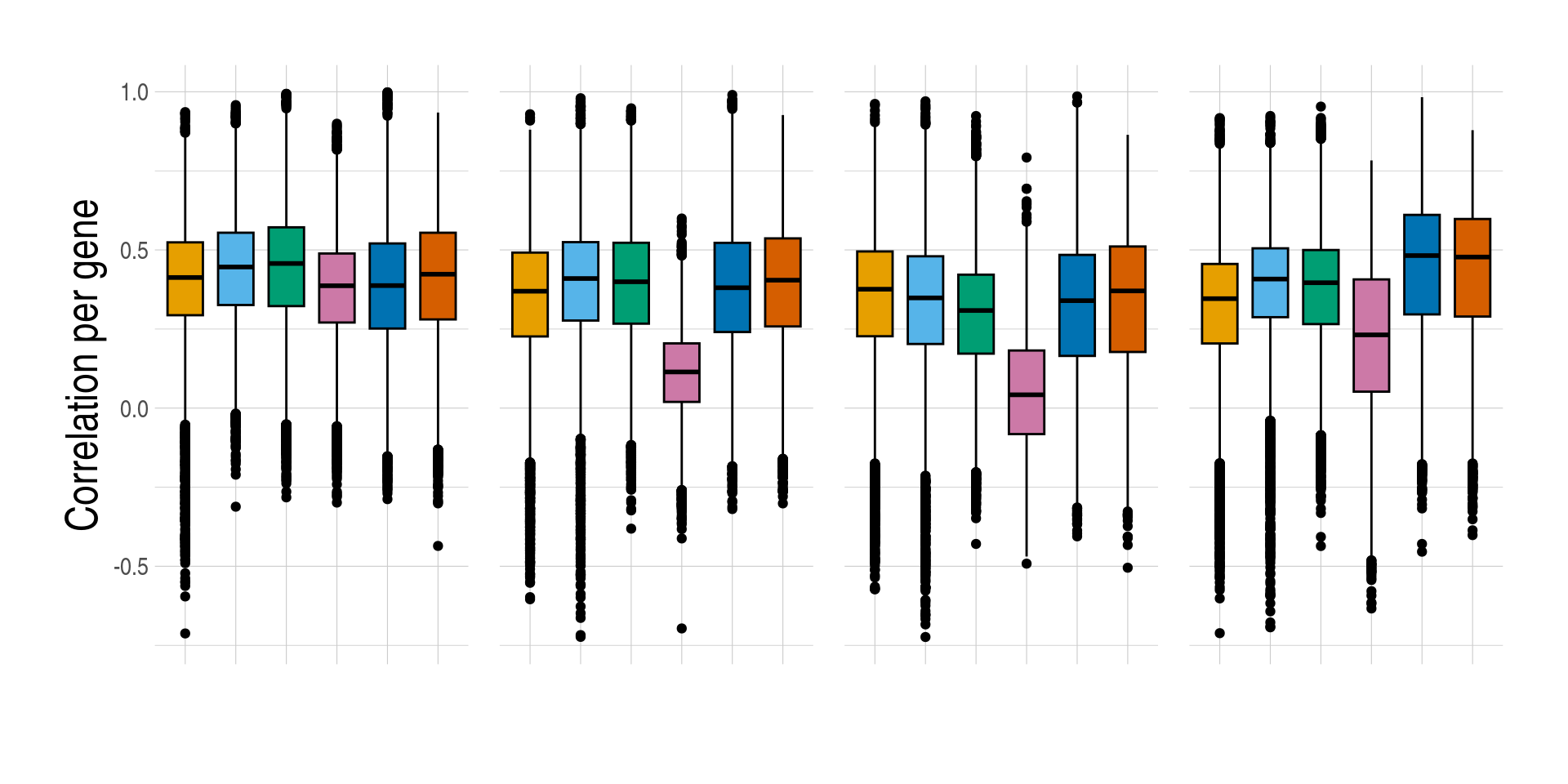
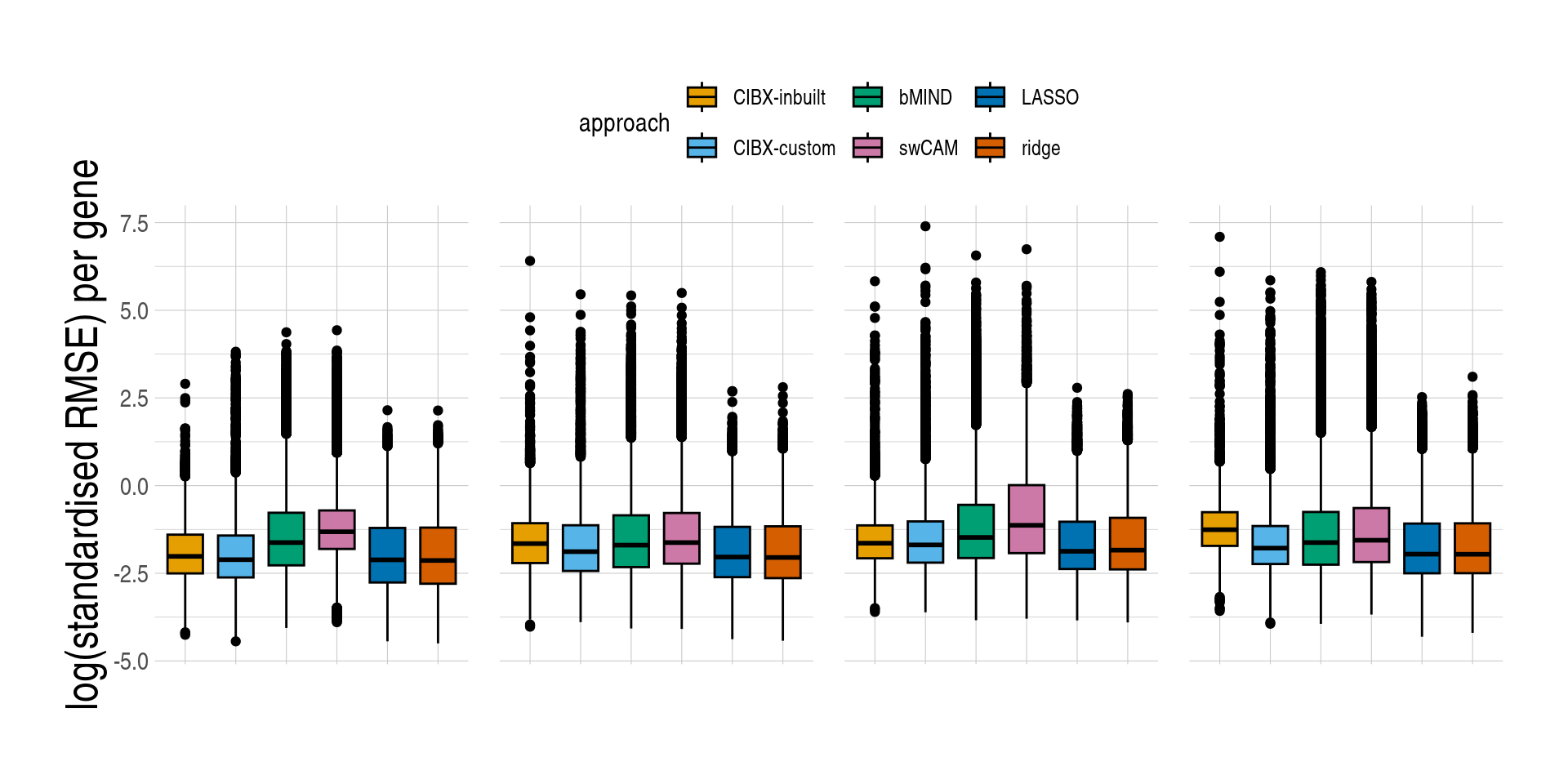
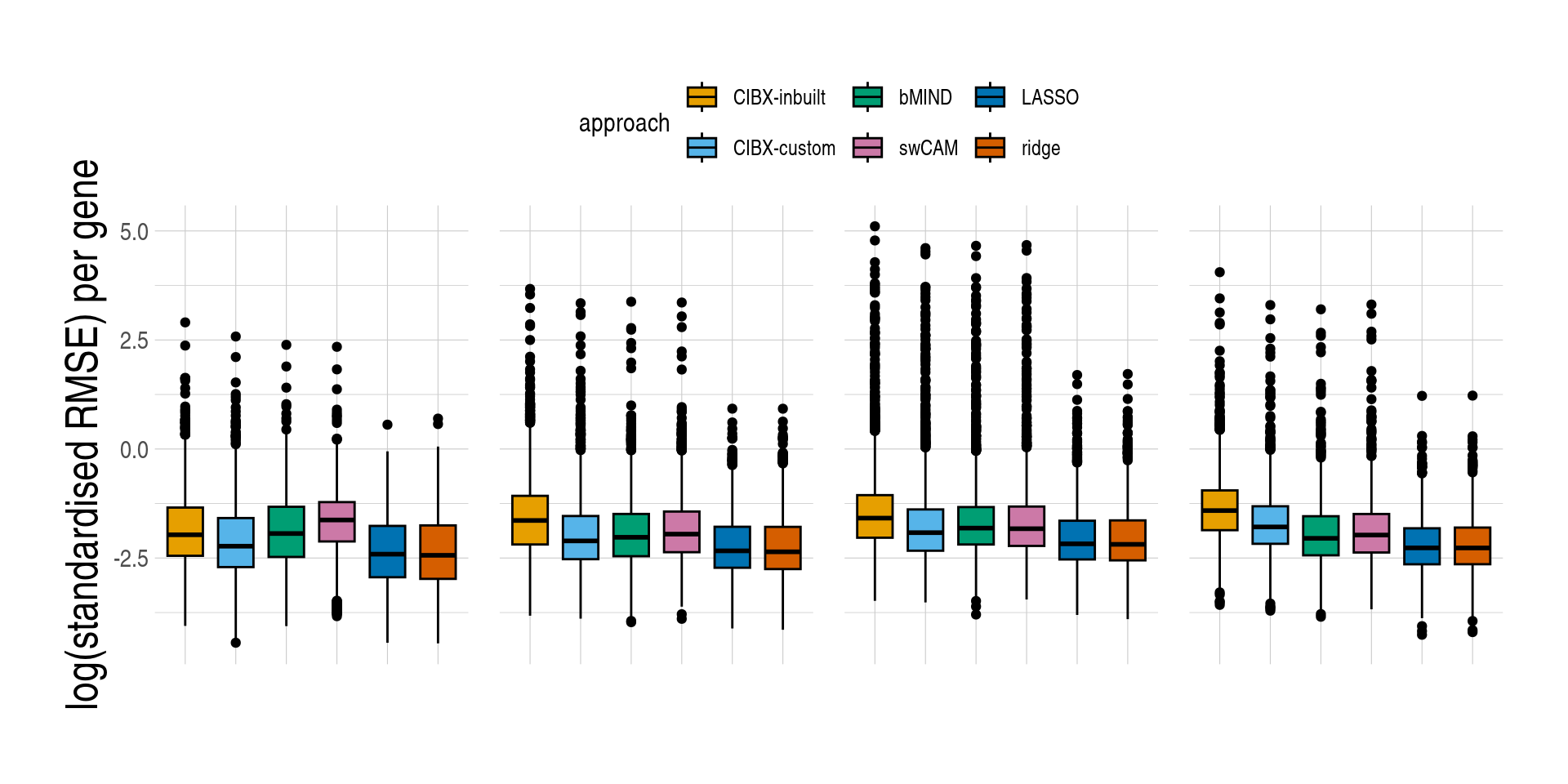
)][, measure_rmse := log(measure_rmse)]- Distributions of prediction accuracy by cell type and approach. Prediction accuracy was evaluated by calculating Pearson correlation and root mean square error (RMSE) between observed and predicted cell-type expression across testing samples for each gene by cell type and approach. Standardised RMSEs: RMSE/average observed expression. The approaches used were: inbuilt - CIBERSORTx expression deconvolution with the inbuilt signature matrix, custom - CIBERSORTx expression deconvolution with a custom signature matrix derived from sorted cell-type expression in training samples, bMIND - bMIND expression deconvolution with flow fractions, swCAM - swCAM deconvolution with flow fractions, and LASSO/RIDGE - expression predicted from regularised multi-response Gaussian models.
Code
measureii <- c("Correlation per gene", "log(standardised RMSE) per gene")
names(measureii) <- c("measure_cor", "measure_rmse")
lapply(seq_along(measureii), function(idx) {
aplt <- ggboxplot(
data = pedaccdsn,
x = "approach",
xlab = "",
y = names(measureii)[idx],
ylab = measureii[idx],
fill = "approach",
palette = appro.cols %>% as.vector(),
facet.by = c("celltype")
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(~celltype, scales = "free")
if (idx == 1) {
aplt <- aplt +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "none",
strip.text = element_blank()
)
} else {
aplt <- aplt +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "top",
strip.text = element_blank()
)
}
aplt
})
Code
fig4_f <- "fig4res.rds"
if (!file.exists(fig4_f)) {
## prepare data to run fig4
myrunfig4 <- run_datprepfig4(
obvexpr = cdata_explst$oexpr,
impvexpr = cdata_explst$iexpr,
tsampct = cdata_explst$trainsamp
)
names(myrunfig4)
## dge & roc
fig4res <- run_fig4(myrunfig4)
names(fig4res)
saveRDS(fig4res, file = fig4_f)
}
if (file.exists(fig4_f)) {
fig4res <- readRDS(fig4_f)
}1.5 Figure 5 , Differential gene expression (DGE) recovery
Code
fig4_f <- "fig4rundata.rds"
if (!file.exists(fig4_f)) {
## main: different gene set; common: same genes per cell across methods
## impvexpr_all <- list(main = expr_ori, common = expr_scenarios)
impvexpr_all <- list(
main = cdata_explst$iexpr,
common = cdata_explst$icexpr
)
## TRUE for dichotomous; FALSE for continuous
dcht_pheno <- c(TRUE, FALSE)
names(dcht_pheno) <- c("dcht", "cntn")
numCores <- length(expr_order)
numCores
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
aucall <- foreach(
j = seq_along(impvexpr_all),
.final = function(x) setNames(x, names(impvexpr_all))
) %:%
foreach(
i = dcht_pheno,
.final = function(x) setNames(x, names(dcht_pheno))
) %dopar% {
observy <- cdata_explst$oexpr
train_samps_ct <- cdata_explst$trainsamp
sampdata <- cdata_explst$phenodata
dat <- run_datprepfig4(
obvexpr = observy,
impvexpr = impvexpr_all[[j]],
tsampct = train_samps_ct,
dich = i
)
## expr ~ pheno
res1 <- run_fig4(datprlst = dat, truesig = 0.05)
## expr~ sex + pheno
res2 <- run_fig4(
datprlst = dat, truesig = 0.05,
covdata = sampdata, covid = "sample", covar = "sex"
)
list(nocov = res1, covin = res2)
}
stopCluster(cl)
mychk <- unlist(aucall, recursive = FALSE) %>%
unlist(., recursive = FALSE) %>%
lapply(., "[[", "aucres") %>%
lapply(., function(datin) {
datin[, ":="(
celltype = factor(celltype, levels = sctorder),
approach = factor(approach, levels = expr_order))]
datin
}) %>%
rbindlist(l = ., idcol = "scenarios") %>%
.[, pheno := gsub("^main\\.|^common\\.", "", scenarios)] %>%
.[, scenario := str_extract(scenarios, "main|common")]
saveRDS(mychk, file = fig4_f)
} else {
mychk <- readRDS(fig4_f)
}Code
pheno.order <- CJ(c("dcht", "cntn"), c("nocov", "covin"), sorted = FALSE)[, paste(V1, V2, sep = ".")]
pheno.order
[1] "dcht.nocov" "dcht.covin" "cntn.nocov" "cntn.covin"
mychk[, pheno := factor(
pheno,
levels = pheno.order,
labels = c("dichPheno", "dichPheno+sex", "contPheno", "contPheno+sex")
)]
ggboxplot(
data = mychk[scenario == "main", ],
x = "approach", y = "auc", ylab = "AUC",
palette = appro.cols,
alpha = 0.35, fill = "approach",
add = c("jitter"),
add.params = list(shape = 21, size = 2.5, alpha = 1)
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(pheno ~ celltype, scales = "free") +
labs(fill = NULL) +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "top",
strip.text.y.right = element_text(angle = 0),
legend.text = element_text(size = rel(1))
) +
rremove("xlab")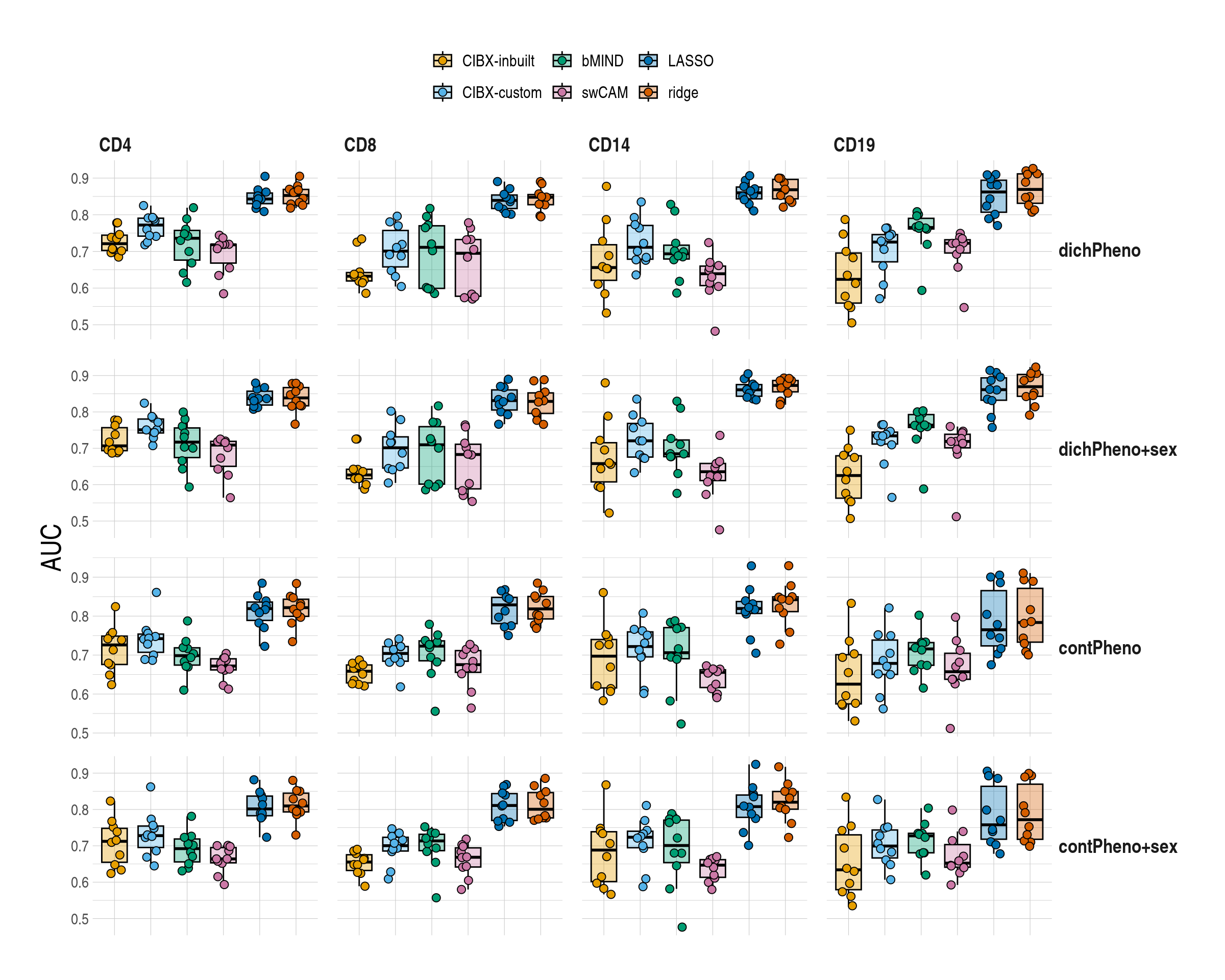
Code
mychk_tbl <- copy(mychk)
mychk_tbl[, .N, by = c("scenarios", "celltype", "approach")]
scenarios celltype approach N
1: main.dcht.nocov CD4 inbuilt 10
2: main.dcht.nocov CD4 custom 10
3: main.dcht.nocov CD4 bMIND 10
4: main.dcht.nocov CD4 swCAM 10
5: main.dcht.nocov CD4 LASSO 10
---
188: common.cntn.covin CD19 custom 10
189: common.cntn.covin CD19 bMIND 10
190: common.cntn.covin CD19 swCAM 10
191: common.cntn.covin CD19 LASSO 10
192: common.cntn.covin CD19 RIDGE 10
tbl_summary <- mychk_tbl[, list(
Q50 = median(auc) %>% round(., 2),
Q25 = quantile(auc, probs = 0.25) %>% round(., 2),
Q75 = quantile(auc, probs = 0.75) %>% round(., 2)
),
by = c("scenarios", "celltype", "approach")
][
, V1 := paste0(Q50, " (", Q25, "-", Q75, ")")
]
tbl_summary[, pheno := gsub("^main\\.|^common\\.", "", scenarios)]
tbl_summary[, scenario := str_extract(scenarios, "main|common")]
tbl_res <- dcast(
data = tbl_summary, pheno + celltype ~ scenario + approach,
value.var = "V1"
)
setDF(tbl_res, rownames = paste0(tbl_res$pheno, tbl_res$celltype))
pheno.order <- CJ(c("dcht", "cntn"), c("nocov", "covin"), sorted = FALSE)[, paste(V1, V2, sep = ".")]
cols.order <- CJ(c("main", "common"), V2 = expr_order, sorted = FALSE)[, paste(V1, V2, sep = "_")]
rows.order <- CJ(pheno.order, sctorder, sorted = FALSE)[, paste0(pheno.order, sctorder)]
tbl_out <- tbl_res[rows.order, c("celltype", grep("main", cols.order, value = TRUE))]
names(tbl_out) <- gsub("main_|common_", "", names(tbl_out))
kbl(
x = tbl_out, row.names = FALSE,
caption = "Median AUC (Q25-Q75) across 10 simulated phentoypes per cell by apparoch. **No. predicted genes vary with cell type and approaches.**"
) %>%
kable_paper("striped", full_width = FALSE) %>%
pack_rows("dichotomous pheno", 1, 4,
label_row_css = "text-align: left;"
) %>%
pack_rows("dichotomous pheno + sex", 5, 8) %>%
pack_rows("continous pheno", 9, 12) %>%
pack_rows("continous pheno + sex", 13, 16) %>%
column_spec(column = 6:7, bold = T)| celltype | inbuilt | custom | bMIND | swCAM | LASSO | RIDGE |
|---|---|---|---|---|---|---|
| dichotomous pheno | ||||||
| CD4 | 0.72 (0.7-0.74) | 0.77 (0.74-0.79) | 0.74 (0.68-0.76) | 0.72 (0.67-0.72) | 0.84 (0.83-0.86) | 0.85 (0.83-0.87) |
| CD8 | 0.63 (0.62-0.64) | 0.7 (0.66-0.76) | 0.71 (0.6-0.77) | 0.69 (0.58-0.73) | 0.84 (0.82-0.85) | 0.85 (0.83-0.86) |
| CD14 | 0.66 (0.62-0.72) | 0.71 (0.68-0.77) | 0.69 (0.68-0.72) | 0.64 (0.61-0.66) | 0.86 (0.84-0.88) | 0.87 (0.84-0.9) |
| CD19 | 0.62 (0.56-0.7) | 0.73 (0.67-0.75) | 0.77 (0.76-0.79) | 0.72 (0.7-0.73) | 0.86 (0.81-0.89) | 0.87 (0.83-0.91) |
| dichotomous pheno + sex | ||||||
| CD4 | 0.71 (0.69-0.76) | 0.75 (0.74-0.78) | 0.72 (0.67-0.76) | 0.71 (0.65-0.72) | 0.84 (0.82-0.86) | 0.84 (0.82-0.87) |
| CD8 | 0.63 (0.62-0.64) | 0.7 (0.65-0.73) | 0.71 (0.6-0.76) | 0.68 (0.59-0.71) | 0.83 (0.81-0.86) | 0.83 (0.8-0.85) |
| CD14 | 0.66 (0.61-0.72) | 0.72 (0.68-0.77) | 0.68 (0.68-0.72) | 0.64 (0.61-0.66) | 0.86 (0.84-0.88) | 0.87 (0.86-0.89) |
| CD19 | 0.63 (0.56-0.68) | 0.73 (0.71-0.74) | 0.76 (0.76-0.79) | 0.72 (0.7-0.74) | 0.86 (0.83-0.89) | 0.87 (0.84-0.9) |
| continous pheno | ||||||
| CD4 | 0.73 (0.68-0.75) | 0.74 (0.71-0.75) | 0.7 (0.67-0.72) | 0.67 (0.66-0.69) | 0.82 (0.79-0.84) | 0.82 (0.8-0.84) |
| CD8 | 0.66 (0.63-0.67) | 0.7 (0.68-0.72) | 0.72 (0.69-0.74) | 0.68 (0.66-0.71) | 0.83 (0.78-0.85) | 0.82 (0.79-0.85) |
| CD14 | 0.7 (0.62-0.74) | 0.72 (0.7-0.76) | 0.71 (0.69-0.77) | 0.66 (0.62-0.66) | 0.82 (0.81-0.84) | 0.84 (0.81-0.85) |
| CD19 | 0.63 (0.57-0.7) | 0.68 (0.65-0.74) | 0.72 (0.67-0.73) | 0.66 (0.64-0.7) | 0.76 (0.72-0.87) | 0.78 (0.73-0.87) |
| continous pheno + sex | ||||||
| CD4 | 0.71 (0.65-0.75) | 0.73 (0.7-0.75) | 0.69 (0.66-0.72) | 0.66 (0.65-0.7) | 0.8 (0.78-0.84) | 0.81 (0.79-0.84) |
| CD8 | 0.66 (0.63-0.68) | 0.7 (0.69-0.72) | 0.71 (0.69-0.73) | 0.67 (0.64-0.69) | 0.81 (0.77-0.84) | 0.8 (0.78-0.85) |
| CD14 | 0.69 (0.6-0.74) | 0.72 (0.69-0.74) | 0.7 (0.65-0.77) | 0.65 (0.61-0.66) | 0.81 (0.78-0.84) | 0.82 (0.8-0.85) |
| CD19 | 0.63 (0.58-0.73) | 0.7 (0.67-0.74) | 0.73 (0.68-0.73) | 0.65 (0.64-0.7) | 0.76 (0.72-0.86) | 0.77 (0.72-0.87) |
1.6 Fig6, simdata
Code
simdata_dir <- file.path(here::here(), "simDataRes")
simdata_dir
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/simDataRes"
sim_ids <- list.dirs(file.path(simdata_dir, "deconvInput"),
full.names = FALSE, recursive = FALSE
)
sim_ids
[1] "1Msim_Combatdat.160.1.100" "1Msim_Combatdat.160.1.25"
[3] "1Msim_Combatdat.160.1.50" "1Msim_Combatdat.160.1.75"
[5] "1Msim_Combatdat.160.2.100" "1Msim_Combatdat.160.2.25"
[7] "1Msim_Combatdat.160.2.50" "1Msim_Combatdat.160.2.75"
[9] "1Msim_Combatdat.160.3.100" "1Msim_Combatdat.160.3.25"
[11] "1Msim_Combatdat.160.3.50" "1Msim_Combatdat.160.3.75"
[13] "1Msim_dat.160.1.100" "1Msim_dat.160.1.25"
[15] "1Msim_dat.160.1.50" "1Msim_dat.160.1.75"
[17] "1Msim_dat.160.2.100" "1Msim_dat.160.2.25"
[19] "1Msim_dat.160.2.50" "1Msim_dat.160.2.75"
[21] "1Msim_dat.160.3.100" "1Msim_dat.160.3.25"
[23] "1Msim_dat.160.3.50" "1Msim_dat.160.3.75"
names(sim_ids) <- sim_ids
sim_ids <- str_sort(sim_ids, numeric = TRUE)
sim_ids
[1] "1Msim_Combatdat.160.1.25" "1Msim_Combatdat.160.1.50"
[3] "1Msim_Combatdat.160.1.75" "1Msim_Combatdat.160.1.100"
[5] "1Msim_Combatdat.160.2.25" "1Msim_Combatdat.160.2.50"
[7] "1Msim_Combatdat.160.2.75" "1Msim_Combatdat.160.2.100"
[9] "1Msim_Combatdat.160.3.25" "1Msim_Combatdat.160.3.50"
[11] "1Msim_Combatdat.160.3.75" "1Msim_Combatdat.160.3.100"
[13] "1Msim_dat.160.1.25" "1Msim_dat.160.1.50"
[15] "1Msim_dat.160.1.75" "1Msim_dat.160.1.100"
[17] "1Msim_dat.160.2.25" "1Msim_dat.160.2.50"
[19] "1Msim_dat.160.2.75" "1Msim_dat.160.2.100"
[21] "1Msim_dat.160.3.25" "1Msim_dat.160.3.50"
[23] "1Msim_dat.160.3.75" "1Msim_dat.160.3.100"
simdata_dir
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/simDataRes"
noimpvgene <- mclapply(sim_ids, function(runid) {
resin <- prep_dat(rid = runid, res_dir = simdata_dir)
## main
mainres <- sapply(unlist(resin$iexpr, recursive = FALSE), nrow)
## common
commres <- sapply(unlist(resin$icexpr, recursive = FALSE), nrow)
data.table(
runid = runid,
appro = tstrsplit(names(mainres), "\\.", keep = 1L) %>% unlist(),
celltype = tstrsplit(names(mainres), "\\.", keep = 2L) %>% unlist(),
mainNo = mainres,
commNo = commres[match(names(mainres), names(commres))]
)
}, mc.cores = 6) %>% rbindlist()Code
noimpvgene[, ":="(
rungrp = str_remove(runid, pattern = "\\.[[:digit:]]+$"),
trainPer = tstrsplit(runid, "\\.", keep = 4L) %>%
unlist(),
CombatYN = ifelse(str_detect(runid, "Combatdat"), "Y", "N")
)]
simnopredtbl <- dcast(data = noimpvgene, celltype + runid ~ appro, value.var = "mainNo")
simnopredtbl[, celltype := factor(celltype, sctorder)]
simnopredtbl[, runid := factor(runid, levels = unique(runid) %>% str_sort(., numeric = TRUE))]
setorder(simnopredtbl, celltype, runid)
setcolorder(simnopredtbl, neworder = c("runid", "celltype", expr_order))
r_idx <- apply(simnopredtbl[, ..expr_order], 1, function(x) any(x < 100)) %>% which()
kbl(x = simnopredtbl, caption = "No.predicted genes", booktabs = TRUE) %>%
kable_paper("striped", full_width = F) %>%
row_spec(row = r_idx, bold = TRUE)| runid | celltype | inbuilt | custom | bMIND | swCAM | LASSO | RIDGE |
|---|---|---|---|---|---|---|---|
| 1Msim_Combatdat.160.1.25 | CD4 | 3353 | 4776 | 11986 | 11987 | 11982 | 11646 |
| 1Msim_Combatdat.160.1.50 | CD4 | 3272 | 4665 | 11993 | 11994 | 11913 | 11913 |
| 1Msim_Combatdat.160.1.75 | CD4 | 3619 | 4924 | 11995 | 11996 | 11910 | 11994 |
| 1Msim_Combatdat.160.1.100 | CD4 | 3519 | 4714 | 12005 | 12006 | 12004 | 11931 |
| 1Msim_Combatdat.160.2.25 | CD4 | 2108 | 4347 | 11961 | 11975 | 11926 | 11972 |
| 1Msim_Combatdat.160.2.50 | CD4 | 2853 | 4284 | 11977 | 11981 | 11979 | 11979 |
| 1Msim_Combatdat.160.2.75 | CD4 | 2716 | 4356 | 11979 | 11981 | 11979 | 11914 |
| 1Msim_Combatdat.160.2.100 | CD4 | 2703 | 4295 | 11973 | 11974 | 11972 | 11972 |
| 1Msim_Combatdat.160.3.25 | CD4 | 3183 | 6160 | 11982 | 11983 | 11879 | 11707 |
| 1Msim_Combatdat.160.3.50 | CD4 | 3502 | 5601 | 11971 | 11976 | 11942 | 11974 |
| 1Msim_Combatdat.160.3.75 | CD4 | 3465 | 5535 | 11976 | 11981 | 11979 | 11935 |
| 1Msim_Combatdat.160.3.100 | CD4 | 3698 | 5559 | 12461 | 12464 | 12461 | 12416 |
| 1Msim_dat.160.1.25 | CD4 | 1128 | 4388 | 11977 | 11977 | 11917 | 11898 |
| 1Msim_dat.160.1.50 | CD4 | 1053 | 4791 | 11981 | 11981 | 11955 | 11907 |
| 1Msim_dat.160.1.75 | CD4 | 1215 | 4319 | 11992 | 11992 | 11990 | 11990 |
| 1Msim_dat.160.1.100 | CD4 | 1212 | 4301 | 11996 | 11996 | 11994 | 11994 |
| 1Msim_dat.160.2.25 | CD4 | 1085 | 4112 | 11983 | 11984 | 11886 | 11945 |
| 1Msim_dat.160.2.50 | CD4 | 1512 | 4191 | 11998 | 11999 | 11997 | 11997 |
| 1Msim_dat.160.2.75 | CD4 | 1384 | 4237 | 11997 | 11997 | 11995 | 11981 |
| 1Msim_dat.160.2.100 | CD4 | 1373 | 4917 | 11986 | 11986 | 11974 | 11974 |
| 1Msim_dat.160.3.25 | CD4 | 973 | 4691 | 11964 | 11966 | 11905 | 11855 |
| 1Msim_dat.160.3.50 | CD4 | 1076 | 5242 | 11960 | 11964 | 11933 | 11928 |
| 1Msim_dat.160.3.75 | CD4 | 1160 | 5215 | 11963 | 11964 | 11962 | 11949 |
| 1Msim_dat.160.3.100 | CD4 | 2026 | 5225 | 12463 | 12463 | 12460 | 12460 |
| 1Msim_Combatdat.160.1.25 | CD8 | 2595 | 4292 | 11975 | 11966 | 11924 | 11723 |
| 1Msim_Combatdat.160.1.50 | CD8 | 2569 | 4295 | 11978 | 11966 | 11986 | 11986 |
| 1Msim_Combatdat.160.1.75 | CD8 | 2737 | 4137 | 11986 | 11991 | 11939 | 11939 |
| 1Msim_Combatdat.160.1.100 | CD8 | 3021 | 4235 | 11997 | 11997 | 11999 | 11999 |
| 1Msim_Combatdat.160.2.25 | CD8 | 2638 | 5067 | 11941 | 11954 | 11886 | 11884 |
| 1Msim_Combatdat.160.2.50 | CD8 | 3300 | 5010 | 11944 | 11978 | 11974 | 11974 |
| 1Msim_Combatdat.160.2.75 | CD8 | 3244 | 4941 | 11953 | 11974 | 11974 | 11899 |
| 1Msim_Combatdat.160.2.100 | CD8 | 3380 | 4850 | 11958 | 11970 | 11968 | 11968 |
| 1Msim_Combatdat.160.3.25 | CD8 | 2626 | 6193 | 11958 | 11982 | 11976 | 11805 |
| 1Msim_Combatdat.160.3.50 | CD8 | 2714 | 5679 | 11941 | 11975 | 11970 | 11970 |
| 1Msim_Combatdat.160.3.75 | CD8 | 2821 | 5603 | 11943 | 11980 | 11975 | 11975 |
| 1Msim_Combatdat.160.3.100 | CD8 | 2960 | 5563 | 12439 | 12461 | 12457 | 12457 |
| 1Msim_dat.160.1.25 | CD8 | 2291 | 4780 | 11964 | 11977 | 11970 | 11970 |
| 1Msim_dat.160.1.50 | CD8 | 2158 | 4107 | 11951 | 11981 | 11953 | 11974 |
| 1Msim_dat.160.1.75 | CD8 | 2142 | 4735 | 11979 | 11992 | 11985 | 11985 |
| 1Msim_dat.160.1.100 | CD8 | 2063 | 4695 | 11978 | 11995 | 11989 | 11972 |
| 1Msim_dat.160.2.25 | CD8 | 1984 | 5045 | 11929 | 11984 | 11884 | 11855 |
| 1Msim_dat.160.2.50 | CD8 | 1789 | 5240 | 11945 | 11999 | 11975 | 11975 |
| 1Msim_dat.160.2.75 | CD8 | 1804 | 5091 | 11966 | 11995 | 11965 | 11971 |
| 1Msim_dat.160.2.100 | CD8 | 1903 | 4493 | 11972 | 11986 | 11979 | 11964 |
| 1Msim_dat.160.3.25 | CD8 | 1823 | 5582 | 11939 | 11966 | 11960 | 11855 |
| 1Msim_dat.160.3.50 | CD8 | 1766 | 4154 | 11924 | 11961 | 11960 | 11960 |
| 1Msim_dat.160.3.75 | CD8 | 1765 | 4215 | 11912 | 11964 | 11919 | 11960 |
| 1Msim_dat.160.3.100 | CD8 | 1958 | 4293 | 12432 | 12461 | 12455 | 12455 |
| 1Msim_Combatdat.160.1.25 | CD14 | 2730 | 6680 | 11956 | 11981 | 11985 | 11956 |
| 1Msim_Combatdat.160.1.50 | CD14 | 3545 | 6799 | 11976 | 11993 | 11993 | 11993 |
| 1Msim_Combatdat.160.1.75 | CD14 | 3604 | 6772 | 11981 | 11992 | 11990 | 11981 |
| 1Msim_Combatdat.160.1.100 | CD14 | 3767 | 6838 | 11992 | 12000 | 12005 | 12005 |
| 1Msim_Combatdat.160.2.25 | CD14 | 3098 | 6798 | 11969 | 11935 | 11923 | 11933 |
| 1Msim_Combatdat.160.2.50 | CD14 | 3426 | 6805 | 11976 | 11959 | 11961 | 11904 |
| 1Msim_Combatdat.160.2.75 | CD14 | 3376 | 6808 | 11975 | 11973 | 11980 | 11962 |
| 1Msim_Combatdat.160.2.100 | CD14 | 3382 | 6909 | 11971 | 11972 | 11937 | 11919 |
| 1Msim_Combatdat.160.3.25 | CD14 | 2845 | 4604 | 11977 | 11931 | 11924 | 11957 |
| 1Msim_Combatdat.160.3.50 | CD14 | 2760 | 4937 | 11970 | 11926 | 11955 | 11965 |
| 1Msim_Combatdat.160.3.75 | CD14 | 2845 | 4999 | 11976 | 11917 | 11980 | 11980 |
| 1Msim_Combatdat.160.3.100 | CD14 | 3529 | 5265 | 12459 | 12433 | 12462 | 12462 |
| 1Msim_dat.160.1.25 | CD14 | 3875 | 3467 | 11950 | 11977 | 11930 | 11976 |
| 1Msim_dat.160.1.50 | CD14 | 4268 | 2135 | 11966 | 11981 | 11980 | 11980 |
| 1Msim_dat.160.1.75 | CD14 | 4333 | 3987 | 11973 | 11992 | 11991 | 11979 |
| 1Msim_dat.160.1.100 | CD14 | 4386 | 3989 | 11985 | 11996 | 11986 | 11995 |
| 1Msim_dat.160.2.25 | CD14 | 3770 | 4915 | 11976 | 11980 | 11967 | 11983 |
| 1Msim_dat.160.2.50 | CD14 | 4077 | 4553 | 11991 | 11999 | 11970 | 11979 |
| 1Msim_dat.160.2.75 | CD14 | 4213 | 4750 | 11995 | 11996 | 11988 | 11989 |
| 1Msim_dat.160.2.100 | CD14 | 4172 | 2656 | 11983 | 11986 | 11985 | 11985 |
| 1Msim_dat.160.3.25 | CD14 | 4353 | 3073 | 11960 | 11966 | 11940 | 11941 |
| 1Msim_dat.160.3.50 | CD14 | 4395 | 4008 | 11962 | 11964 | 11964 | 11948 |
| 1Msim_dat.160.3.75 | CD14 | 4267 | 4040 | 11961 | 11964 | 11949 | 11944 |
| 1Msim_dat.160.3.100 | CD14 | 4457 | 4207 | 12461 | 12459 | 12455 | 12455 |
| 1Msim_Combatdat.160.1.25 | CD19 | 8 | 982 | 11924 | 10073 | 11922 | 11919 |
| 1Msim_Combatdat.160.1.50 | CD19 | 55 | 968 | 11863 | 9961 | 11935 | 11906 |
| 1Msim_Combatdat.160.1.75 | CD19 | 14 | 892 | 11863 | 9945 | 11946 | 11946 |
| 1Msim_Combatdat.160.1.100 | CD19 | 51 | 927 | 11879 | 10128 | 11952 | 11952 |
| 1Msim_Combatdat.160.2.25 | CD19 | 1 | 1059 | 11948 | 11381 | 11871 | 11893 |
| 1Msim_Combatdat.160.2.50 | CD19 | 1 | 1095 | 11935 | 10023 | 11926 | 11904 |
| 1Msim_Combatdat.160.2.75 | CD19 | 32 | 1018 | 11923 | 10167 | 11917 | 11927 |
| 1Msim_Combatdat.160.2.100 | CD19 | 57 | 945 | 11895 | 8760 | 11910 | 11898 |
| 1Msim_Combatdat.160.3.25 | CD19 | 6 | 86 | 11942 | 11653 | 11913 | 11932 |
| 1Msim_Combatdat.160.3.50 | CD19 | 13 | 1298 | 11915 | 11364 | 11904 | 11928 |
| 1Msim_Combatdat.160.3.75 | CD19 | 33 | 1342 | 11928 | 11311 | 11931 | 11901 |
| 1Msim_Combatdat.160.3.100 | CD19 | 46 | 1476 | 12374 | 11824 | 12377 | 12377 |
| 1Msim_dat.160.1.25 | CD19 | 1198 | 2589 | 11958 | 10689 | 11914 | 11906 |
| 1Msim_dat.160.1.50 | CD19 | 1144 | 3409 | 11923 | 11183 | 11919 | 11914 |
| 1Msim_dat.160.1.75 | CD19 | 1396 | 2252 | 11938 | 11159 | 11933 | 11930 |
| 1Msim_dat.160.1.100 | CD19 | 1956 | 2323 | 11945 | 11578 | 11940 | 11940 |
| 1Msim_dat.160.2.25 | CD19 | 1823 | 1475 | 11939 | 11887 | 11909 | 11924 |
| 1Msim_dat.160.2.50 | CD19 | 2647 | 1478 | 11914 | 11992 | 11941 | 11934 |
| 1Msim_dat.160.2.75 | CD19 | 2875 | 1391 | 11907 | 11961 | 11943 | 11943 |
| 1Msim_dat.160.2.100 | CD19 | 2783 | 2870 | 11880 | 11973 | 11927 | 11923 |
| 1Msim_dat.160.3.25 | CD19 | 1052 | 1821 | 11954 | 11370 | 11891 | 11906 |
| 1Msim_dat.160.3.50 | CD19 | 787 | 2100 | 11939 | 10372 | 11908 | 11907 |
| 1Msim_dat.160.3.75 | CD19 | 474 | 2085 | 11939 | 10721 | 11906 | 11913 |
| 1Msim_dat.160.3.100 | CD19 | 2310 | 2124 | 12401 | 10990 | 12375 | 12366 |
Code
simres_f <- "simres_chunksize.rds"
if (!file.exists(simres_f)) {
sim_pen_chunks <- lapply(seq_along(sim_ids), function(idx) {
chunk_files <- file.path(
simdata_dir,
paste0("PenalisedInput/", sim_ids[idx])
) %>% list.files(
pattern = "*_chunk[[:digit:]]+.rds",
full.names = TRUE
)
chunk_celltypes <- str_extract(
basename(chunk_files),
"CD4|CD8|CD14|CD19"
)
chunk_s <- sapply(chunk_files, function(c_f) {
readRDS(c_f)[["yyinput"]] %>% ncol()
})
data.table(celltype = chunk_celltypes, chunksize = chunk_s)
}) %>%
set_names(x = ., value = sim_ids) %>%
rbindlist(., idcol = "simid")
saveRDS(object = sim_pen_chunks, file = simres_f)
}
if (file.exists(simres_f)) {
sim_pen_chunks <- readRDS(simres_f)
}
## no.chunks by cell type across pesudobulk data
sim_pen_chunks[,
{
Nchunks <- .N
sizesmin <- min(chunksize)
sizesmax <- max(chunksize)
list(Nchunks, sizesmin, sizesmax)
},
by = c("celltype")
][match(sctorder, celltype), ]
celltype Nchunks sizesmin sizesmax
1: CD4 1358 4 500
2: CD8 1306 8 498
3: CD14 1533 3 499
4: CD19 1180 2 497Code
simdata_dir
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/simDataRes"
simres_f <- "simres_all.rds"
if (!file.exists(simres_f)) {
simres_all <- lapply(sim_ids, function(runid) {
message(runid)
datin <- prep_dat(rid = runid, res_dir = simdata_dir)
commonexpr <- datin$icexpr
## 0 genes common across methods
if (any(sapply(datin$icexpr[[1]], nrow) == 0)) {
k_ct <- which(sapply(datin$icexpr[[1]], nrow) != 0) %>%
names(datin$icexpr[[1]])[.]
commonexpr <- lapply(datin$icexpr, function(xin) {
xin[k_ct]
})
}
impvexpr_all <- list(
main = datin$iexpr,
common = commonexpr
)
observy <- datin$oexpr
train_samps_ct <- datin$trainsamp
sampdata <- datin$phenodata
## sce1: train+test in [1] & [2]
## sce2: test in [1] & [2]
sce_list <- list(
sce1 = list(tsin = train_samps_ct, testsamp = FALSE),
sce2 = list(tsin = NULL, testsamp = FALSE)
)
numCores <- length(impvexpr_all) * length(sce_list)
numCores
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
aucall <- foreach(
j = seq_along(impvexpr_all),
.final = function(x) setNames(x, names(impvexpr_all))
) %:%
foreach(
i = seq_along(sce_list),
.final = function(x) setNames(x, names(sce_list))
) %dopar% {
tsin <- sce_list[[i]][["tsin"]]
ttin <- sce_list[[i]][["testsamp"]]
dat <- run_datprepfig4(
obvexpr = observy,
impvexpr = impvexpr_all[[j]],
tsampct = tsin,
phenodata = sampdata,
dich = TRUE
)
## expr ~ pheno
res1 <- run_fig4(
datprlst = dat,
truesig = 0.05, testsamp = ttin
)
## expr~ batch + pheno
res2 <- run_fig4(
datprlst = dat, truesig = 0.05,
testsamp = ttin,
covdata = sampdata, covid = "sample",
covar = "batch"
)
list(nocov = res1, covin = res2)
}
stopCluster(cl)
aucall
})
names(simres_all) <- sim_ids
simresmychk <- unlist(simres_all, recursive = FALSE) %>%
unlist(., recursive = FALSE) %>%
unlist(., recursive = FALSE) %>%
lapply(., "[[", "aucres") %>%
rbindlist(l = ., idcol = "runidsce")
saveRDS(simresmychk, file = simres_f)
}
if (file.exists(simres_f)) {
simresmychk <- readRDS(simres_f)
}
simresmychk[, scenario := str_extract(runidsce, "main|common")]
simresmychk[, trainper := tstrsplit(simresmychk$runidsce, "\\.", keep = 4L) %>% unlist()]
simresmychk[, sce := str_extract(runidsce, "sce[1|2|3]")]
simresmychk[, pheno := str_extract(runidsce, "nocov|covin")]
simresmychk[, pheno := ifelse(grepl("Combatdat", runidsce), paste0("Cb", pheno), pheno)]
simresmychk <- simresmychk[pheno != "Cbcovin", ]
simresmychk[, ":="(
celltype = factor(celltype, levels = sctorder),
approach = factor(approach, levels = expr_order),
pheno = factor(pheno,
levels = c("nocov", "covin", "Cbnocov"),
labels = c("cond", "batch+cond", "Combat-Seq\ncond")
)
)]
simresmychk[, trainper := as.numeric(trainper)]
## simresmychk[, trainper := factor(trainper, levels = unique(simresmychk$trainper) %>% str_sort(., numeric = TRUE))]
## n_trainper <- nlevels(simresmychk$trainper)
## n_trainper
## shape_m <- seq(21, length.out = n_trainper) %>%
## set_names(levels(simresmychk$trainper))
simresmychk[, approach := appro_lab[match(simresmychk$approach, names(appro_lab))]]
simresmychk[, approach := factor(approach, appro_lab)]
appro.cols.mod <- appro.cols
names(appro.cols.mod) <- appro_lab
## CIBX-inbuilt imputed < 60 genes for CD19 under Combat-Seq cond
## unstable AUCs, set the rest as NA
simresmychk[pheno == "Combat-Seq\ncond" & celltype == "CD19" & approach == "CIBX-inbuilt", ]
runidsce fakedPCs approach celltype
1: 1Msim_Combatdat.160.1.25.main.sce1.nocov cond CIBX-inbuilt CD19
2: 1Msim_Combatdat.160.1.25.main.sce2.nocov cond CIBX-inbuilt CD19
3: 1Msim_Combatdat.160.1.50.main.sce1.nocov cond CIBX-inbuilt CD19
4: 1Msim_Combatdat.160.1.50.main.sce2.nocov cond CIBX-inbuilt CD19
5: 1Msim_Combatdat.160.1.75.main.sce1.nocov cond CIBX-inbuilt CD19
6: 1Msim_Combatdat.160.1.75.main.sce2.nocov cond CIBX-inbuilt CD19
7: 1Msim_Combatdat.160.1.100.main.sce1.nocov cond CIBX-inbuilt CD19
8: 1Msim_Combatdat.160.1.100.main.sce2.nocov cond CIBX-inbuilt CD19
9: 1Msim_Combatdat.160.2.25.main.sce1.nocov cond CIBX-inbuilt CD19
10: 1Msim_Combatdat.160.2.25.main.sce2.nocov cond CIBX-inbuilt CD19
11: 1Msim_Combatdat.160.2.50.main.sce1.nocov cond CIBX-inbuilt CD19
12: 1Msim_Combatdat.160.2.50.main.sce2.nocov cond CIBX-inbuilt CD19
13: 1Msim_Combatdat.160.2.75.main.sce1.nocov cond CIBX-inbuilt CD19
14: 1Msim_Combatdat.160.2.75.main.sce2.nocov cond CIBX-inbuilt CD19
15: 1Msim_Combatdat.160.2.100.main.sce1.nocov cond CIBX-inbuilt CD19
16: 1Msim_Combatdat.160.2.100.main.sce2.nocov cond CIBX-inbuilt CD19
17: 1Msim_Combatdat.160.3.25.main.sce1.nocov cond CIBX-inbuilt CD19
18: 1Msim_Combatdat.160.3.25.main.sce2.nocov cond CIBX-inbuilt CD19
19: 1Msim_Combatdat.160.3.50.main.sce1.nocov cond CIBX-inbuilt CD19
20: 1Msim_Combatdat.160.3.50.main.sce2.nocov cond CIBX-inbuilt CD19
21: 1Msim_Combatdat.160.3.50.common.sce1.nocov cond CIBX-inbuilt CD19
22: 1Msim_Combatdat.160.3.50.common.sce2.nocov cond CIBX-inbuilt CD19
23: 1Msim_Combatdat.160.3.75.main.sce1.nocov cond CIBX-inbuilt CD19
24: 1Msim_Combatdat.160.3.75.main.sce2.nocov cond CIBX-inbuilt CD19
25: 1Msim_Combatdat.160.3.75.common.sce1.nocov cond CIBX-inbuilt CD19
26: 1Msim_Combatdat.160.3.75.common.sce2.nocov cond CIBX-inbuilt CD19
27: 1Msim_Combatdat.160.3.100.main.sce1.nocov cond CIBX-inbuilt CD19
28: 1Msim_Combatdat.160.3.100.main.sce2.nocov cond CIBX-inbuilt CD19
29: 1Msim_Combatdat.160.3.100.common.sce1.nocov cond CIBX-inbuilt CD19
30: 1Msim_Combatdat.160.3.100.common.sce2.nocov cond CIBX-inbuilt CD19
runidsce fakedPCs approach celltype
auc scenario trainper sce pheno
1: 0.5481005 main 25 sce1 Combat-Seq\ncond
2: NA main 25 sce2 Combat-Seq\ncond
3: 0.5030582 main 50 sce1 Combat-Seq\ncond
4: 0.4960178 main 50 sce2 Combat-Seq\ncond
5: 0.5923738 main 75 sce1 Combat-Seq\ncond
6: 0.9588608 main 75 sce2 Combat-Seq\ncond
7: 0.6417989 main 100 sce1 Combat-Seq\ncond
8: 0.5084310 main 100 sce2 Combat-Seq\ncond
9: NA main 25 sce1 Combat-Seq\ncond
10: NA main 25 sce2 Combat-Seq\ncond
11: NA main 50 sce1 Combat-Seq\ncond
12: NA main 50 sce2 Combat-Seq\ncond
13: 0.6268437 main 75 sce1 Combat-Seq\ncond
14: 0.5205106 main 75 sce2 Combat-Seq\ncond
15: 0.5907763 main 100 sce1 Combat-Seq\ncond
16: 0.5075536 main 100 sce2 Combat-Seq\ncond
17: NA main 25 sce1 Combat-Seq\ncond
18: NA main 25 sce2 Combat-Seq\ncond
19: NA main 50 sce1 Combat-Seq\ncond
20: NA main 50 sce2 Combat-Seq\ncond
21: NA common 50 sce1 Combat-Seq\ncond
22: NA common 50 sce2 Combat-Seq\ncond
23: 0.4792505 main 75 sce1 Combat-Seq\ncond
24: 0.9679359 main 75 sce2 Combat-Seq\ncond
25: NA common 75 sce1 Combat-Seq\ncond
26: NA common 75 sce2 Combat-Seq\ncond
27: 0.5507876 main 100 sce1 Combat-Seq\ncond
28: 0.5317240 main 100 sce2 Combat-Seq\ncond
29: NA common 100 sce1 Combat-Seq\ncond
30: NA common 100 sce2 Combat-Seq\ncond
auc scenario trainper sce pheno
simresmychk[pheno == "Combat-Seq\ncond" & celltype == "CD19" & approach == "CIBX-inbuilt", auc := NA]Code
## main: varying gene sets
## common: gene sets common across approaches
## sce1: train+ test; sce2: test only
simres_pltlist <- lapply(unique(simresmychk$scenario), function(scein) {
lapply(c("sce1", "sce2"), function(xin) {
ggplot(
data = simresmychk[scenario == scein & sce == xin, ],
aes(x = trainper, y = auc, colour = approach)
) +
geom_point( # aes(shape = celltype),
alpha = 0.5, size = 1.5
) +
geom_smooth(
aes(
group = approach,
colour = approach,
linetype = approach,
fill = approach
),
alpha = 0.35, se = FALSE, linewidth = 0.5
) +
scale_colour_manual(values = appro.cols.mod) +
scale_fill_manual(values = appro.cols.mod) +
facet_grid(cols = vars(celltype), rows = vars(pheno)) +
ylab("AUC") +
xlab("Percentage of 80 train samples") +
scale_x_continuous(
breaks = seq(25, 100, by = 25),
labels = scales::label_percent(scale = 1)
) +
used_ggthemes(
strip.text.y.right = element_text(angle = 0),
legend.position = "top"
)
}) %>% set_names(., c("sce1", "sce2"))
}) %>% set_names(., unique(simresmychk$scenario))
simres_pltlist$main$sce1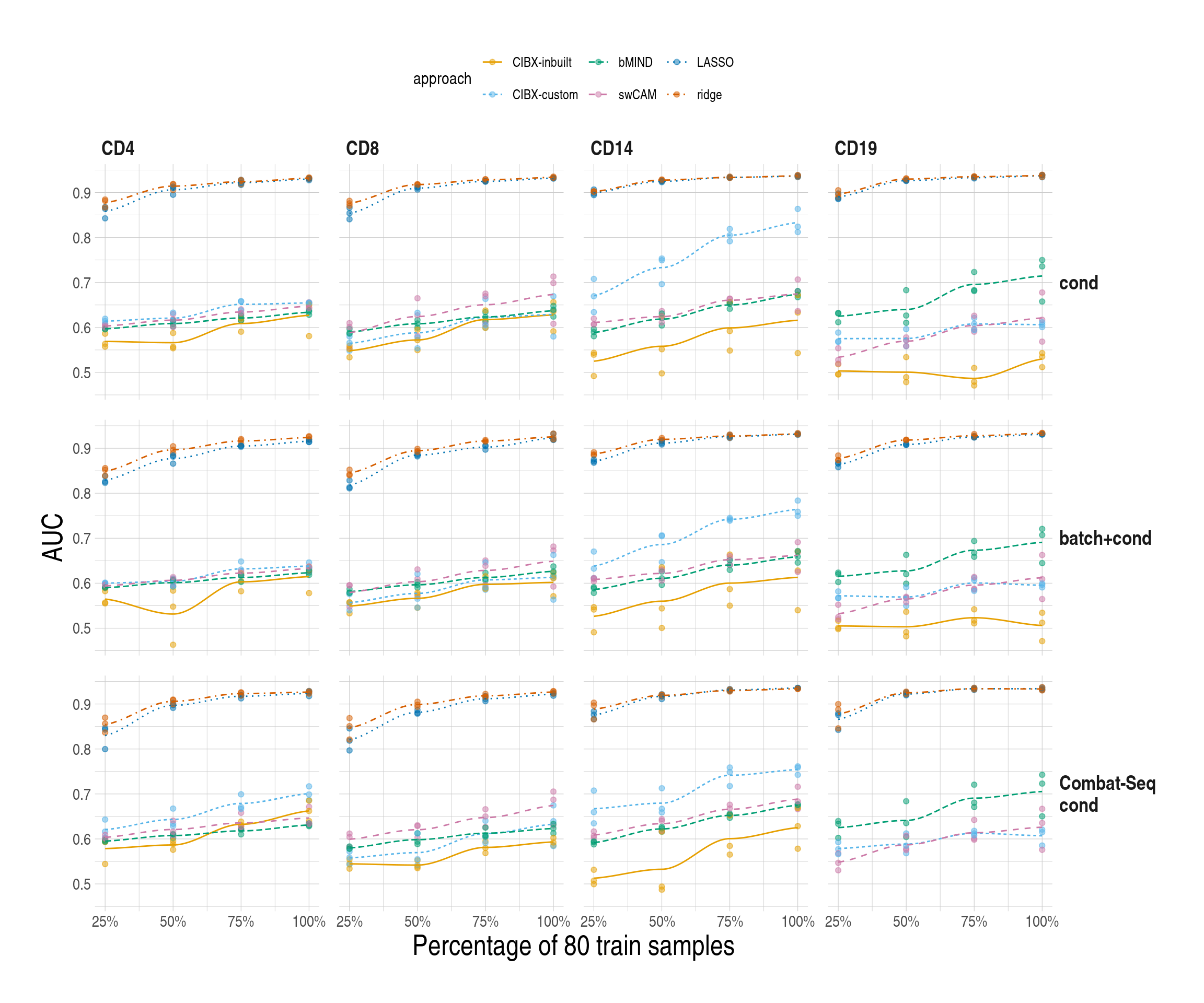
Code
openxlsx::write.xlsx(
list(
"Fig1A" = sampdata,
"Fig2" = pltfract_test,
"Fig2.anno" = cordsn,
"Fig3AB" = pedaccdsn_persub,
"Fig3CD" = pedaccdsn,
"Fig4" = mychk[scenario == "main",
setdiff(names(mychk), c("scenarios", "scenario")),
with = FALSE
],
"Fig5" = simresmychk[
scenario == "main" & sce == "sce1",
.(approach, celltype, auc, trainper, pheno)
]
),
file = "SourceData_mainFigures.xlsx",
overwrite = TRUE
)
figS6 <- mychk[scenario == "common",
setdiff(names(mychk), c("scenarios", "scenario")),
with = FALSE
]
figS8 <- simresmychk[
scenario == "common" & sce == "sce1",
.(approach, celltype, auc, trainper, pheno)
]2 Supplementary material
2.1 SupFig1, Overlap of predicted genes by approach
- Overlap of predicted genes by CIBERSORTx using inbuilt (inbuilt) and our custom signatures (custom), bMIND, swCAM, LASSO and RIDGE. Predicted genes were defined as those with variations in expression across subjects. For each panel (cell type), the right bar plot indicates the numbers of predicted genes (No.Pred.Genes) by approach, and the top bar plot demonstrates No.Pred.Genes common in different combinations of approaches (black dots), but not in the grey-dot approaches, if present.
Code
col_pals <- cols4all::c4a("miscs.okabe", n = 4)
expr_scenarios <- cdata_explst$iexpr
stopifnot(names(expr_scenarios) == expr_order)
names(expr_scenarios) <- appro_lab
m_list <- lapply(seq_along(sctorder), function(idx) {
ct <- sctorder[idx]
Ocom <- lapply(expr_scenarios, function(dat) {
rownames(dat[[ct]])
})
m1_mod <- make_comb_mat(Ocom, mode = "distinct")
m1 <- m1_mod
m1
})
names(m_list) <- sctorder
figS1 <- lapply(seq_along(sctorder), function(idx) {
ct <- sctorder[idx]
Ocom <- lapply(expr_scenarios, function(dat) {
rownames(dat[[ct]])
})
max.n <- sapply(Ocom, length) %>% max()
for (n in seq_along(Ocom)) {
length(Ocom[[n]]) <- max.n
}
do.call(cbind, Ocom)
})
names(figS1) <- paste0("figS1.", sctorder)
sapply(m_list, comb_size)
m_list <- normalize_comb_mat(m_list)
sapply(m_list, comb_size)
max_set_size <- max(sapply(m_list, set_size))
max_comb_size <- max(sapply(m_list, comb_size))
ht_list <- NULL
for (i in seq_along(m_list)) {
ht_list <- ht_list %v%
UpSet(m_list[[i]],
row_names_side = "left",
row_title = paste0(names(m_list)[i]),
row_title_gp = gpar(fontface = 2),
row_title_rot = 0,
set_order = names(expr_scenarios),
comb_order = NULL,
top_annotation = upset_top_annotation(m_list[[i]],
ylim = c(0, max_comb_size),
gp = gpar(fill = col_pals[2]),
annotation_name_rot = 90,
add_numbers = T, numbers_rot = 0
),
right_annotation = upset_right_annotation(m_list[[i]],
ylim = c(0, max_set_size),
gp = gpar(fill = col_pals[3]),
add_numbers = T
)
)
}
ht_list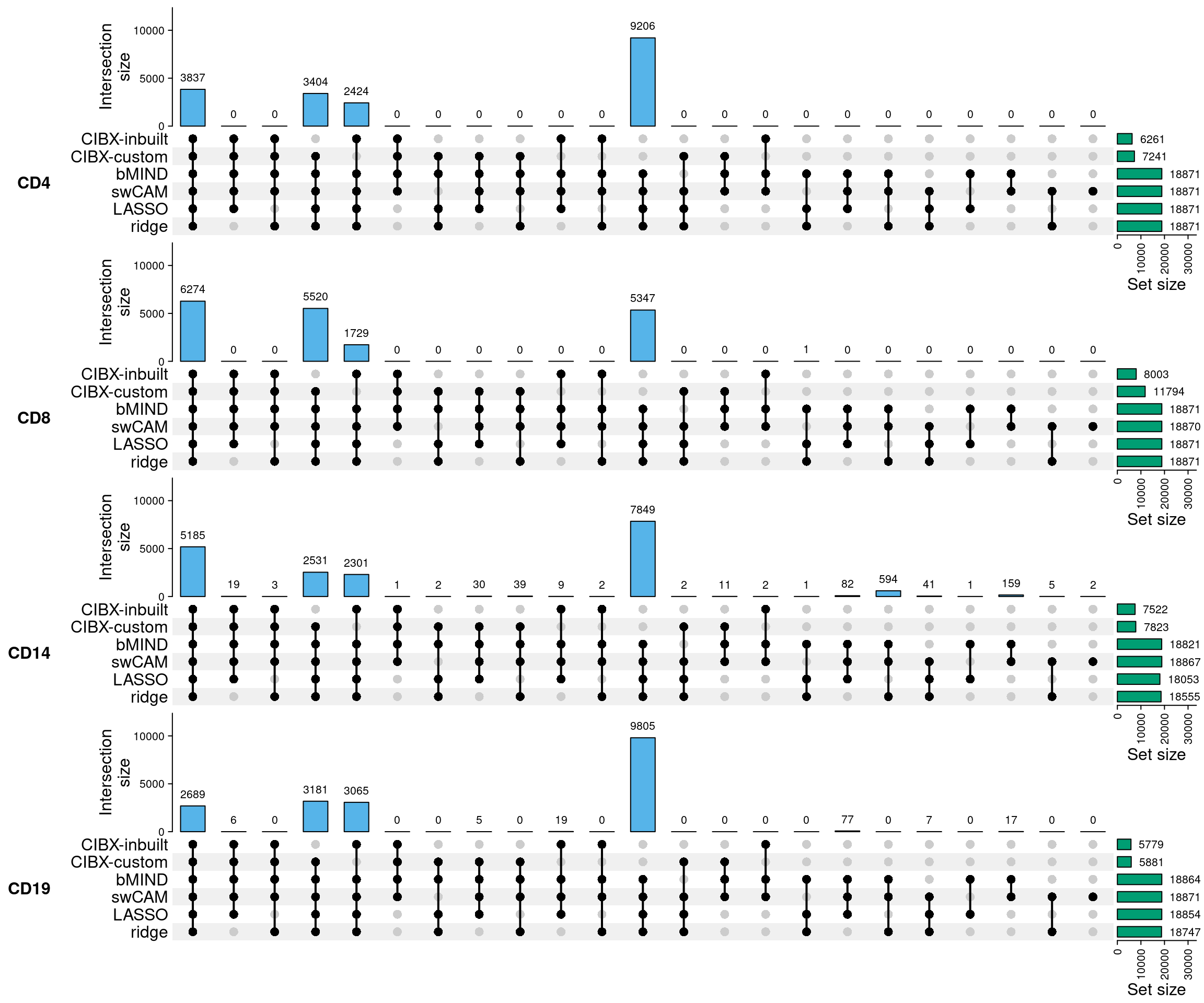
2.2 SupFig2, Correlations per subject stratified by same and different subjects
- Distributions of Pearson correlations (y-axis) between observed and imputed expression across genes from the same/different subjects by cell type and approach. One estimate per subject. inbuilt: CIBERSORTx with the inbuilt signature matrix; custom: CIBERSORTx with a custom signature matrix; bMIND: bMIND with flow fractions; swCAM: swCAM with flow fractions; LASSO/RIDGE: regularised multi-response Gaussian
Code
numCores <- length(expr_order)
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
pedaccdsn_rotpersub <- foreach(j = seq_along(expr_order), .combine = "rbind") %:%
foreach(i = seq(sctorder), .combine = "rbind") %dopar% {
expr_scenarios <- cdata_explst$iexpr
observy <- cdata_explst$oexpr
sce <- expr_order[j]
ctin <- sctorder[i]
expr_in <- expr_scenarios[[sce]][[ctin]]
observ_in <- observy[rownames(expr_in), colnames(expr_in)]
## differnt subjects
rot <- colnames(observ_in)[c(2:ncol(observ_in), 1)]
## correlation across genes per subject
resdsn <- cor(expr_in[, rot], observ_in, method = "pearson") %>% diag()
## rmse across genes per subject
mse1 <- apply((observ_in - expr_in[, rot])^2, 2, mean) %>% sqrt()
stopifnot(names(resdsn) == names(mse1))
## another baseline as observed expression in different subjects
abaseline <- cor(observ_in, observ_in[, rot]) %>% diag()
dsn1 <- data.table(
approach = sce, celltype = ctin,
measure_cor = resdsn,
measure_rmse = mse1,
avecor = abaseline
)
dsn1
}
stopCluster(cl)
pedaccdsn_rotpersub[, .N, by = c("approach", "celltype")]
approach celltype N
1: inbuilt CD4 71
2: inbuilt CD8 65
3: inbuilt CD14 52
4: inbuilt CD19 57
5: custom CD4 71
6: custom CD8 65
7: custom CD14 52
8: custom CD19 57
9: bMIND CD4 71
10: bMIND CD8 65
11: bMIND CD14 52
12: bMIND CD19 57
13: swCAM CD4 71
14: swCAM CD8 65
15: swCAM CD14 52
16: swCAM CD19 57
17: LASSO CD4 71
18: LASSO CD8 65
19: LASSO CD14 52
20: LASSO CD19 57
21: RIDGE CD4 71
22: RIDGE CD8 65
23: RIDGE CD14 52
24: RIDGE CD19 57
approach celltype N
pedaccdsn_rotpersub[, ":="(
approach = factor(approach, levels = expr_order),
celltype = factor(celltype, levels = sctorder)
)]
ped_a <- pedaccdsn_rotpersub[, .(approach, celltype, measure_cor)][
, sce := "diff"
]
ped_b <- pedaccdsn_rotpersub[, .(approach, celltype, avecor)][
, sce := "ObsDiff"
]
setnames(ped_b, old = "avecor", new = "measure_cor")
pedaccdsn_persub[, sce := "same"]
list(pedaccdsn_persub, ped_a, ped_b) %>%
rbindlist(., fill = TRUE) %>%
.[, sce := factor(sce, levels = c("same", "diff", "ObsDiff"))] %>%
ggboxplot(
data = ., x = "celltype", y = "measure_cor",
fill = "sce",
facet.by = "approach",
panel.labs = list(approach = appro_lab),
ylab = "Correlation",
palette = "jco",
ggtheme = used_ggthemes(
legend.position = "top",
legend.text = element_text(size = rel(1.2))
)
) +
labs(fill = "Subject") +
rremove("xlab")
figS2 <- rbindlist(list(pedaccdsn_persub, ped_a, ped_b), fill = TRUE) %>%
.[, sce := factor(sce, levels = c("same", "diff", "ObsDiff"))]
figS2[, (c("measure_rmse")) := NULL]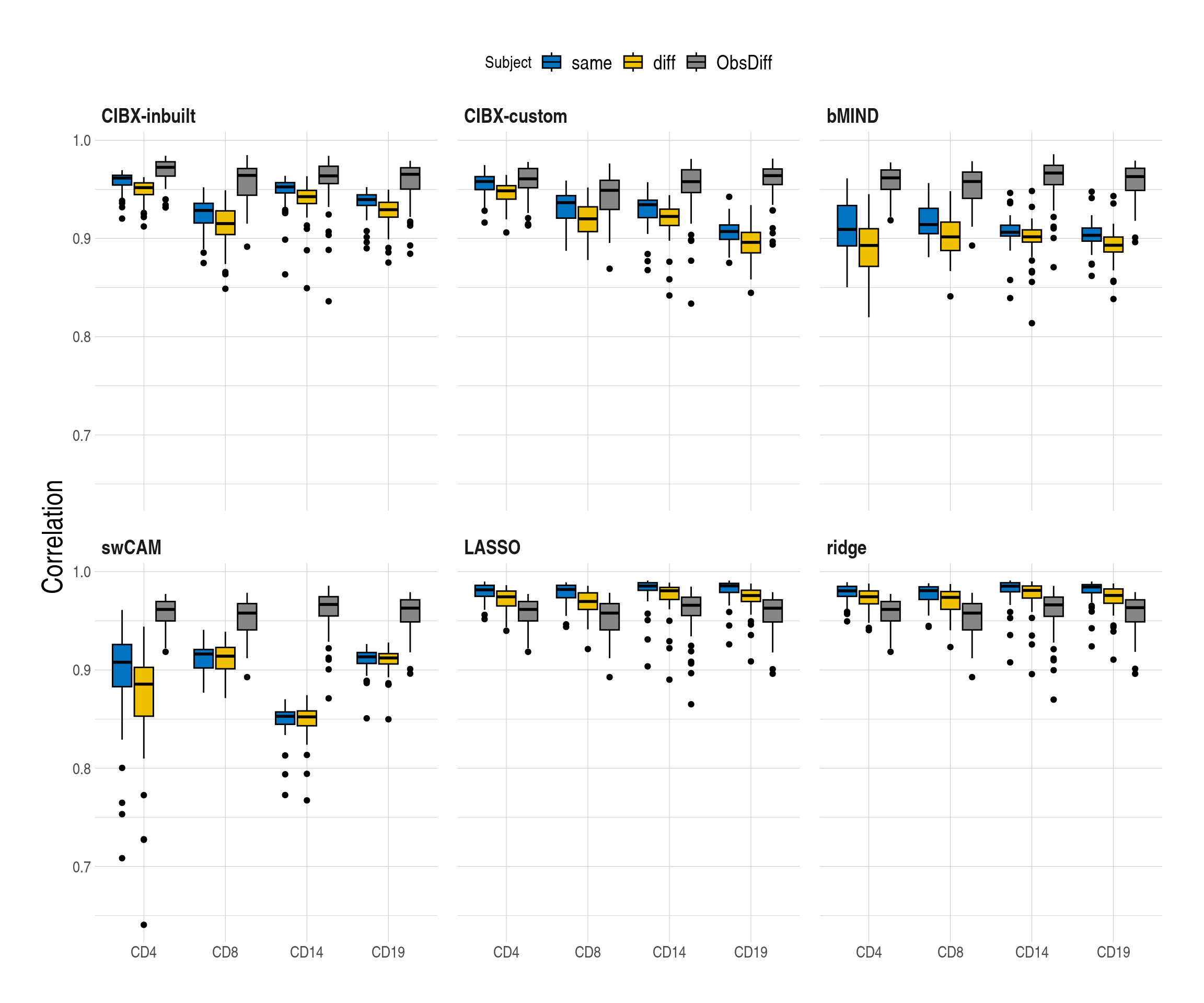
2.3 SupFig3, Common gene sets-CLUSTER data
Code
## expression from approaches
cdata_explst <- prep_dat(
rid = "cluster",
res_dir = data_dir,
cluster = TRUE
)
gcommon <- lapply(sctorder, function(ctin) {
lapply(cdata_explst$icexpr, "[[", ctin) %>%
lapply(., rownames) %>%
Reduce(f = intersect, x = .)
})
names(gcommon) <- sctorder
sapply(gcommon, length)
CD4 CD8 CD14 CD19
3837 6274 5185 2689 2.3.1 correlation & rmse per subject
Code
### predicted accuracy
observy <- cdata_explst$oexpr
expr_scenarios <- cdata_explst$icexpr
numCores <- length(expr_order)
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
pedaccdsn_persub <- foreach(i = expr_order, .combine = "rbind") %dopar% {
run_fig3v(
obvexpr = observy, impvexpr = expr_scenarios,
sce = i, ct = sctorder, persub = TRUE
) %>%
rbindlist()
}
stopCluster(cl)
pedaccdsn_persub[, .N, by = c("approach", "celltype")]
approach celltype N
1: inbuilt CD4 71
2: inbuilt CD8 65
3: inbuilt CD14 52
4: inbuilt CD19 57
5: custom CD4 71
6: custom CD8 65
7: custom CD14 52
8: custom CD19 57
9: bMIND CD4 71
10: bMIND CD8 65
11: bMIND CD14 52
12: bMIND CD19 57
13: swCAM CD4 71
14: swCAM CD8 65
15: swCAM CD14 52
16: swCAM CD19 57
17: LASSO CD4 71
18: LASSO CD8 65
19: LASSO CD14 52
20: LASSO CD19 57
21: RIDGE CD4 71
22: RIDGE CD8 65
23: RIDGE CD14 52
24: RIDGE CD19 57
approach celltype N
pedaccdsn_persub[, ":="(
approach = factor(approach, levels = expr_order),
celltype = factor(celltype, levels = sctorder)
)][, measure_rmse := log(measure_rmse)]
measureii <- c("Correlation per subject", "log(RMSE) per subject")
names(measureii) <- c("measure_cor", "measure_rmse")
lapply(seq_along(measureii), function(idx) {
bplt <- ggboxplot(
data = pedaccdsn_persub,
x = "approach",
xlab = "",
y = names(measureii)[idx],
ylab = measureii[idx],
fill = "approach",
palette = appro.cols,
facet.by = c("celltype")
) +
facet_grid(~celltype, scales = "free")
if (idx == 1) {
bplt <- bplt +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "none"
)
} else {
bplt <- bplt +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "none",
strip.text = element_blank()
)
}
bplt
})
[[1]]
[[2]]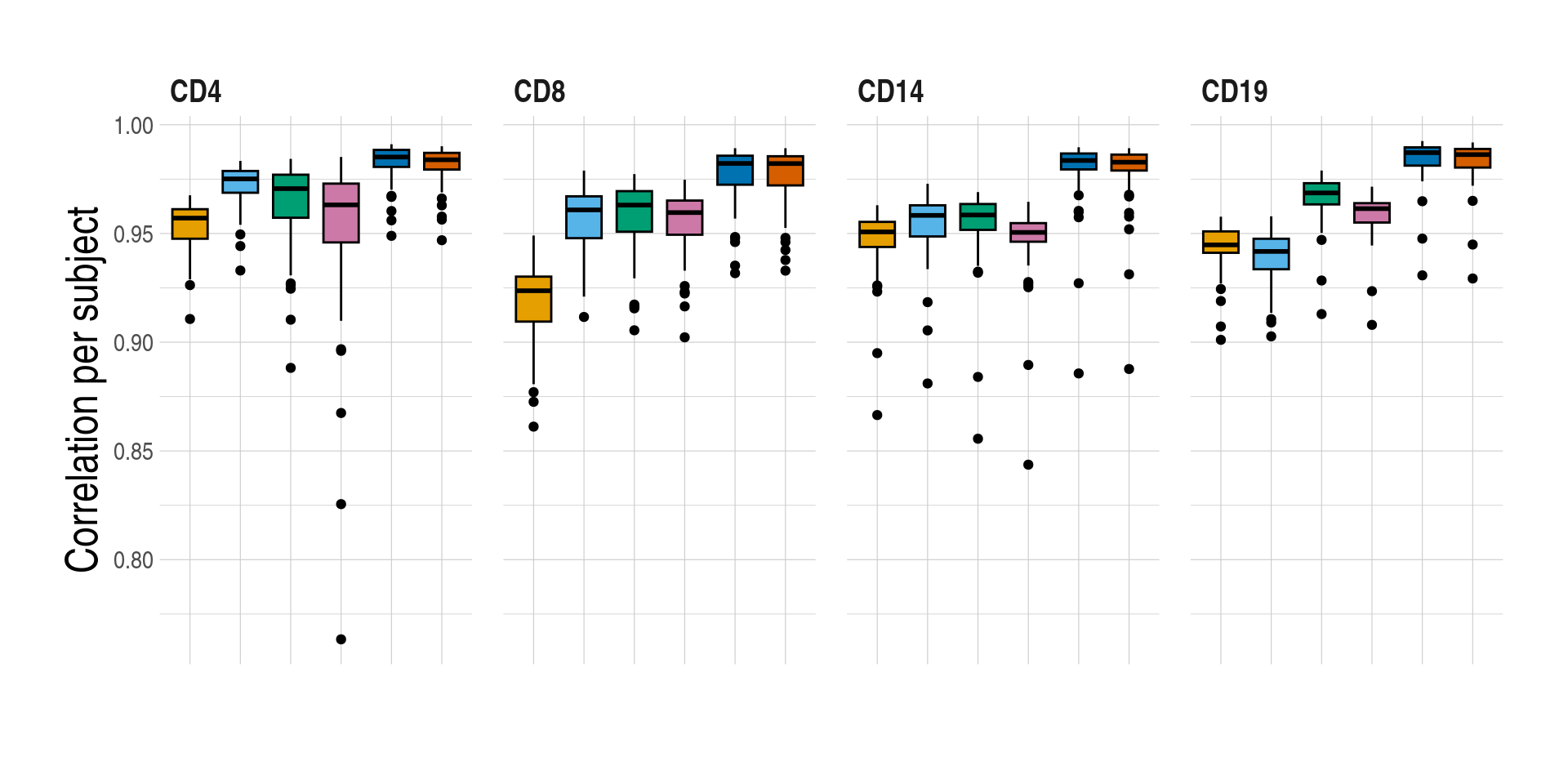
Distributions of prediction accuracy by cell type and approach. Prediction accuracy was evaluated by calculating Pearson correlation and root mean square error (RMSE) between observed and predicted cell-type expression across genes for the same subjects by cell type and approach. For each cell type, genes common across approaches were used. No. common genes are 3837 for CD4, 6274 for CD8, 5185 for CD14, and 2689 for CD19, respectively. The approaches used were: inbuilt - CIBERSORTx expression deconvolution with the inbuilt signature matrix, custom - CIBERSORTx expression deconvolution with a custom signature matrix derived from sorted cell-type expression in training samples, bMIND - bMIND expression deconvolution with flow fractions, swCAM - swCAM deconvolution with flow fractions, and LASSO/RIDGE - expression predicted from regularised multi-response Gaussian models.
2.3.2 correlation & rmse per gene
Code
numCores <- length(expr_order)
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
pedaccdsn <- foreach(i = expr_order, .combine = "rbind") %dopar% {
run_fig3v(
obvexpr = observy, impvexpr = expr_scenarios,
sce = i, ct = sctorder, persub = FALSE
) %>%
rbindlist()
}
stopCluster(cl)
pedaccdsn[, .N, by = c("approach", "celltype")]
approach celltype N
1: inbuilt CD4 3837
2: inbuilt CD8 6274
3: inbuilt CD14 5185
4: inbuilt CD19 2689
5: custom CD4 3837
6: custom CD8 6274
7: custom CD14 5185
8: custom CD19 2689
9: bMIND CD4 3837
10: bMIND CD8 6274
11: bMIND CD14 5185
12: bMIND CD19 2689
13: swCAM CD4 3837
14: swCAM CD8 6274
15: swCAM CD14 5185
16: swCAM CD19 2689
17: LASSO CD4 3837
18: LASSO CD8 6274
19: LASSO CD14 5185
20: LASSO CD19 2689
21: RIDGE CD4 3837
22: RIDGE CD8 6274
23: RIDGE CD14 5185
24: RIDGE CD19 2689
approach celltype N
pedaccdsn[, ":="(
approach = factor(approach, levels = expr_order),
celltype = factor(celltype, levels = sctorder)
)][, measure_rmse := log(measure_rmse)]Code
measureii <- c("Correlation per gene", "log(standardised RMSE) per gene")
names(measureii) <- c("measure_cor", "measure_rmse")
lapply(seq_along(measureii), function(idx) {
aplt <- ggboxplot(
data = pedaccdsn,
x = "approach",
xlab = "",
y = names(measureii)[idx],
ylab = measureii[idx],
fill = "approach",
palette = appro.cols,
facet.by = c("celltype")
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(~celltype, scales = "free")
if (idx == 1) {
aplt <- aplt + used_ggthemes(
axis.text.x = element_blank(),
legend.position = "none",
strip.text = element_blank()
)
} else {
aplt <- aplt + used_ggthemes(
axis.text.x = element_blank(),
legend.position = "top",
strip.text = element_blank()
)
}
aplt
})
[[1]]
[[2]]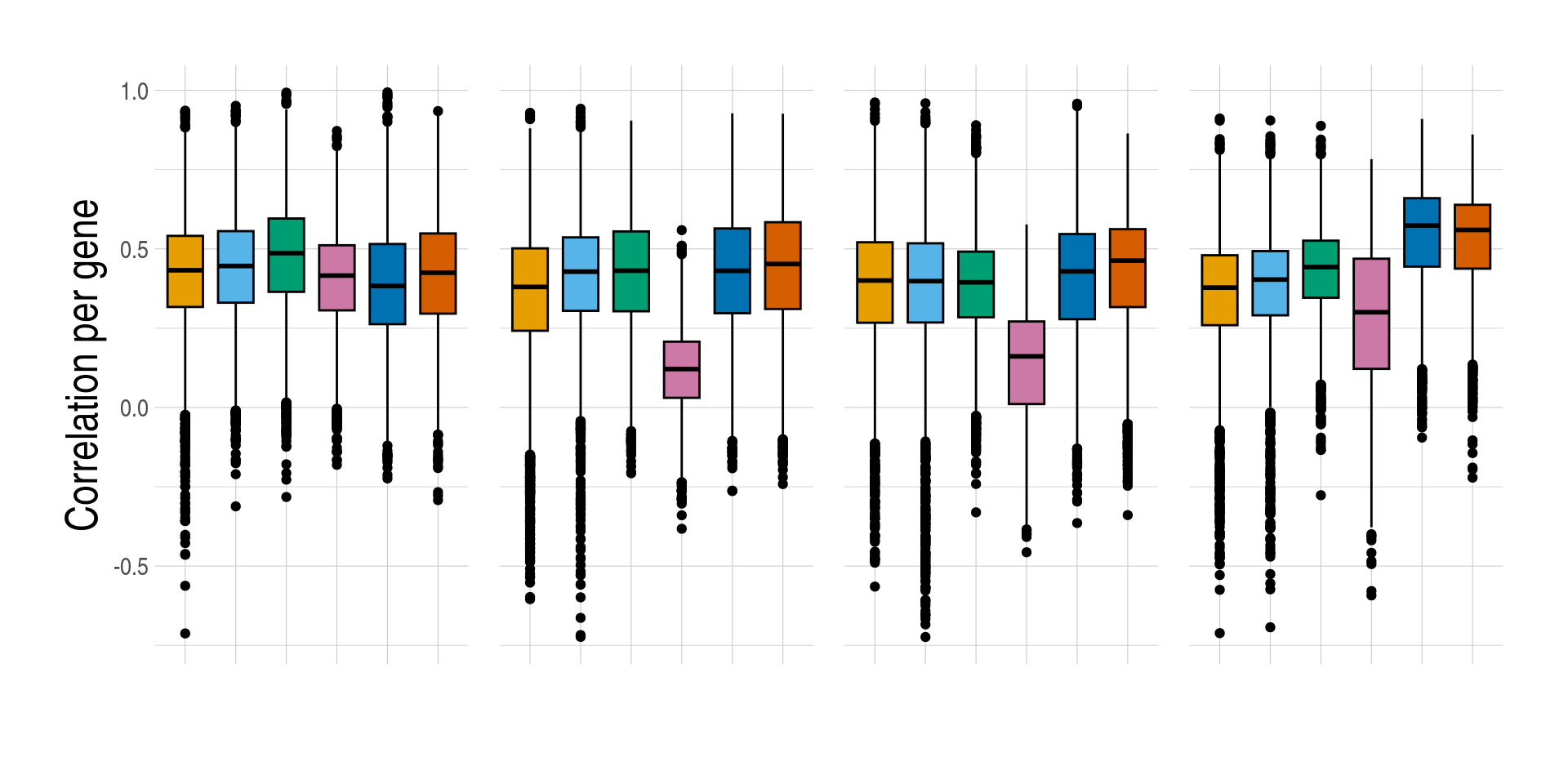
Distributions of prediction accuracy by cell type and approach. Prediction accuracy was evaluated by calculating Pearson correlation and root mean square error (RMSE) between observed and predicted cell-type expression across testing samples for each gene by cell type and approach. For each cell type, genes common across approaches were used. No. common genes are 3837 for CD4, 6274 for CD8, 5185 for CD14, and 2689 for CD19, respectively. Standardised RMSEs: RMSE/average observed expression. The approaches used were: inbuilt - CIBERSORTx expression deconvolution with the inbuilt signature matrix, custom - CIBERSORTx expression deconvolution with a custom signature matrix derived from sorted cell-type expression in training samples, bMIND - bMIND expression deconvolution with flow fractions, swCAM - swCAM deconvolution with flow fractions, and LASSO/RIDGE - expression predicted from regularised multi-response Gaussian models.
Code
figS3ab <- pedaccdsn_persub
figS3cd <- pedaccdsn2.4 SupFig4,5-DGE recovery
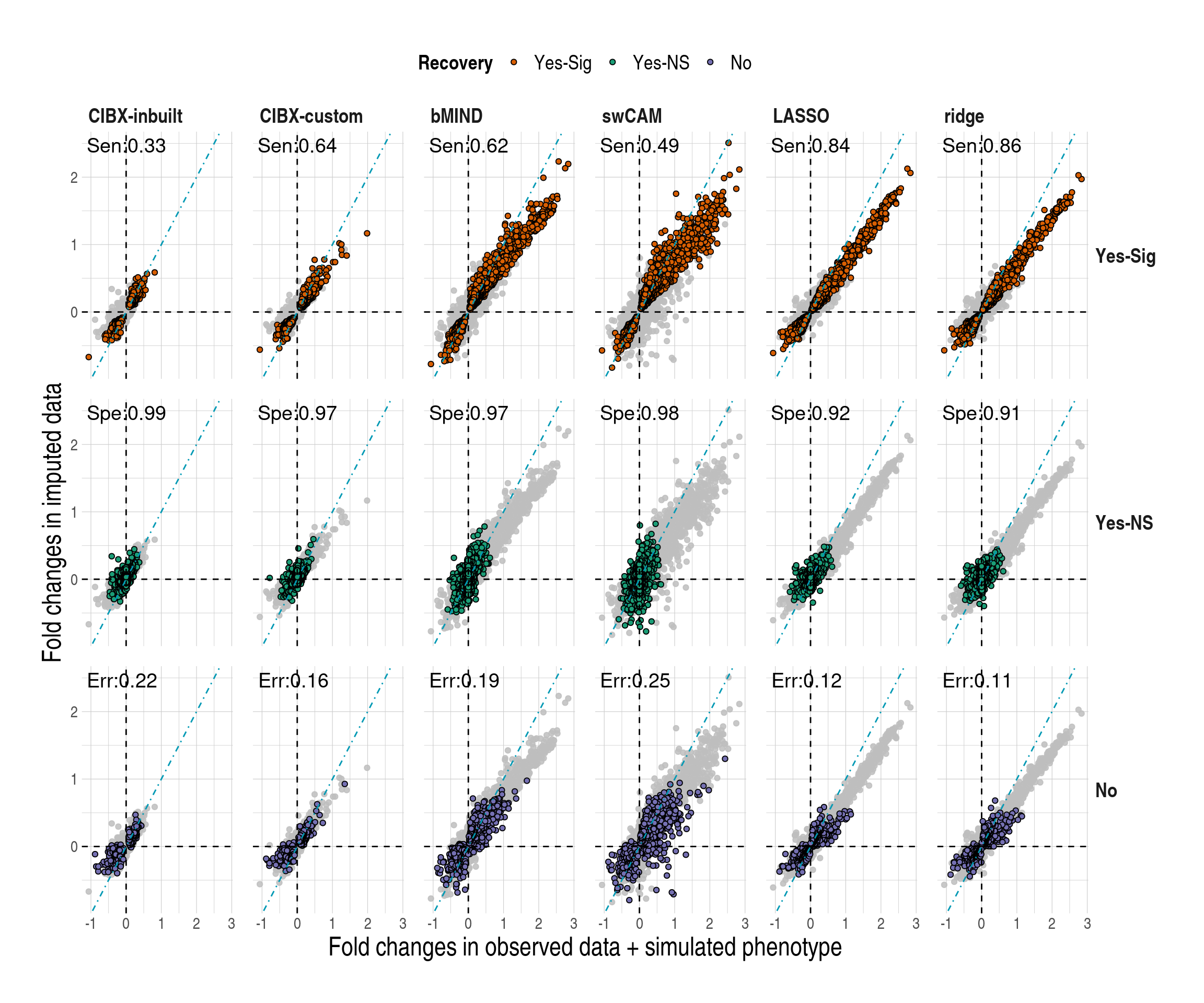
- Comparisons of log\(_2\) fold changes in genes between the observed (x-axis) and imputed (y-axis) data by method (column) and by recovery status (row). DGE analysis was carried out using limma based on one of the simulated phenotypes. An FDR of 0.05 was used in both observed and imputed data here, and CD4 DGE recovery is shown. DGE results in each method (column) are the same, with coloured points for genes falling into that category of recovery status (row) and grey points for genes not belonging to the same category. Recovery status: No, Yes-NS, and Yes-Sig. Yes-Sig (sensitivity; Sen): differentially expressed genes in the observed data were also called significance in the imputed data, and the orientations of the effect sizes are the same in both data. Yes-NS (specificity; Spe): genes are called non-significance (NS) in both data. No (error; Err): misclassified genes; Err is calculated as the percentage of misclassified genes to the total number of predicted genes.
Code
degres <- copy(fig4res$dge_res) %>% setDT()
degres[, celltype := factor(celltype, levels = sctorder)]
degres[, approach := factor(approach, levels = expr_order)]
## FDR in true
fdr.thr <- 0.05
## use truecol and callmade
degres[, truecol := "0No"][adj.P.Val < fdr.thr, truecol := "1Yes"]
degres[, callmade := "0Nonsig"][
impvadjp < fdr.thr & sign(t) == sign(impvt),
callmade := "1Yes"
]
degres[, recovery := "No"][truecol == "1Yes" & callmade == "1Yes", recovery := "Yes-Sig"][
truecol == "0No" & callmade == "0Nonsig", recovery := "Yes-NS"
]
## numerator
annotdsn <- degres[, .N, by = c("recovery", "fakedpheno", "approach", "celltype")]
annotdsn1 <- degres[truecol == "0No",
{
N_NS_obs <- .N
list(N_NS_obs)
},
by = c("fakedpheno", "approach", "celltype")
][
degres[truecol == "1Yes",
{
N_SIG_obs <- .N
list(N_SIG_obs)
},
by = c("fakedpheno", "approach", "celltype")
], N_SIG_obs := i.N_SIG_obs,
on = c("fakedpheno", "approach", "celltype")
][is.na(N_SIG_obs), N_SIG_obs := 0][
annotdsn[recovery == "Yes-NS", ], N_recNS := i.N,
on = c("fakedpheno", "approach", "celltype")
][
annotdsn[recovery == "Yes-Sig", ], N_recSIG := i.N,
on = c("fakedpheno", "approach", "celltype")
][is.na(N_recSIG), N_recSIG := 0]
tidelabfun <- function(a, b, group) {
## a1 <- paste0(group, ":", a, "/", b)
a1 <- paste0(group, ":")
if (b != 0) {
c <- round(a / b, 2)
## a1 <- paste0(a1, " (", c, ")")
a1 <- paste0(a1, c)
}
a1
}
annotdsn1[, ":="(
tlab = paste0(
tidelabfun(N_recSIG, N_SIG_obs, "Sig"), "\n",
tidelabfun(N_recNS, N_NS_obs, "NS "), "\n",
tidelabfun(N_recNS + N_recSIG, N_SIG_obs + N_NS_obs, "ALL")
)
),
by = c("fakedpheno", "approach", "celltype")
]
mypaldeg <- cols4all::c4a("brewer.dark2", n = 3)
names(mypaldeg) <- c("Yes-NS", "Yes-Sig", "No")
annotdsn1[, ":="(
tlab = paste0(
tidelabfun(N_recSIG, N_SIG_obs, "Sen"), "\n",
tidelabfun(N_recNS, N_NS_obs, "Spe")
)
),
by = c("fakedpheno", "approach", "celltype")
]
mypaldeg <- cols4all::c4a("brewer.dark2", n = 3)
names(mypaldeg) <- c("Yes-NS", "Yes-Sig", "No")
## PC1 and CD4 subset
pltdsn <- degres[fakedpheno == "PC1" & celltype == "CD4", ]
pltdsn[, recovery := factor(recovery, levels = c("Yes-Sig", "Yes-NS", "No"))]
## summary of Sen & Spe, and plot lab
annodf1 <- pltdsn[, .N, by = c("approach", "recovery")]
annodf2 <- pltdsn[truecol == "1Yes", .N, by = c("approach")][, recovery := "Yes-Sig"]
annodf3 <- pltdsn[truecol == "0No", .N, by = c("approach")][, recovery := "Yes-NS"]
annodf4 <- pltdsn[, .N, by = c("approach")][, recovery := "No"]
annodf1[annodf2, Nt := i.N, on = c("approach", "recovery")]
annodf1[annodf3, Nt := i.N, on = c("approach", "recovery")]
annodf1[annodf4, Nt := i.N, on = c("approach", "recovery")]
annodf1[!is.na(Nt), pper := round(N / Nt, 2)]
annodf1[recovery == "Yes-Sig", tlab := paste0("Sen:", pper)]
annodf1[recovery == "Yes-NS", tlab := paste0("Spe:", pper)]
annodf1[recovery == "No", tlab := paste0("Err:", pper)]
kklab <- levels(pltdsn$recovery)
names(kklab) <- kklab
figS4 <- copy(pltdsn)
figS4.anno <- copy(annodf1)
ggplot(data = pltdsn, aes(x = logFC, y = impvlogFC)) +
geom_point(data = pltdsn %>% dplyr::select(-recovery), color = "grey", alpha = 0.85) +
geom_point(aes(fill = recovery), shape = 21) +
geom_hline(yintercept = 0, linetype = 2, color = "black") +
geom_vline(xintercept = 0, linetype = 2, color = "black") +
geom_abline(
intercept = 0, slope = 1, linetype = 4,
color = ggsci::pal_lancet(alpha = 1)(8)[4]
) +
ggrepel::geom_text_repel(
data = annodf1,
aes(
x = -Inf, y = Inf,
label = tlab
),
hjust = 0, size = 5,
min.segment.length = Inf,
parse = FALSE
) +
scale_fill_manual(values = mypaldeg) +
facet_grid(recovery ~ approach,
labeller = as_labeller(c(appro_lab, kklab))
) +
xlab("Fold changes in observed data + simulated phenotype") +
ylab("Fold changes in imputed data") +
labs(fill = "Recovery") +
used_ggthemes(
legend.position = "top",
axis.title = element_text(size = rel(1.2)),
legend.text = element_text(size = rel(1.2)),
legend.title = element_text(
size = rel(1.2),
face = "bold"
),
strip.text.y.right = element_text(angle = 0)
)
- Receiver operating characteristic (ROC) curves and estimated area under curve (AUC) by cell type and approach based on one simulated phenotype. FDR was fixed at 0.05 in the observed data and varied from 0 to 1 by 0.05 in the imputed data.
Code
###################################################################################
## summarise the ROC curves by approach based on the first simulated phenotypes ##
###################################################################################
## pc1 as example
pc_examp <- "PC1"
rocresall <- fig4res$rocresall
myaucs <- fig4res$aucres[fakedPCs == "PC1", ]
myaucs[, auc := round(auc, 3)]
myaucs[, name := paste(celltype, approach, sep = ".")]
appro.cols.MOD <- appro.cols[match(myaucs$approach, names(appro.cols))]
names(appro.cols.MOD) <- myaucs$name
myaucs[, ":="(
celltype = factor(celltype, levels = sctorder),
approach = factor(approach,
levels = expr_order
)
)]
myrun <- lapply(sctorder, function(ctin) {
aucpltdsn <- lapply(rocresall[[pc_examp]][[ctin]], "[[", "rocres")
names(aucpltdsn) <- paste0(ctin, ".", names(aucpltdsn))
aucpltdsn
})
myrun <- unlist(myrun, recursive = FALSE)
library(pROC)
g.list <- ggroc(myrun, legacy.axes = TRUE)
kklab <- sctorder
names(kklab) <- sctorder
g.list$data$celltype <- tstrsplit(g.list$data$name, "\\.", keep = 1L) %>%
unlist() %>%
factor(., levels = sctorder)
g.list$data$approach <- tstrsplit(g.list$data$name, "\\.", keep = 2L) %>%
unlist() %>%
factor(., levels = expr_order)
g.list +
scale_color_manual(values = appro.cols.MOD) +
facet_grid(celltype ~ approach,
labeller = as_labeller(c(appro_lab, kklab))
) +
geom_text(
data = myaucs,
aes(0.75, 0.25,
label = auc
),
size = 6
) +
geom_abline(
slope = 1, intercept = 0, linetype = 2,
colour = cols4all::c4a_na("okabe")
) +
ylab("Sensitivity") +
xlab("1-Specificity") +
used_ggthemes(
legend.position = "none",
strip.text.y.right = element_text(angle = 0)
)
figS5 <- g.list$data
figS5.anno <- myaucs[, .(approach, celltype, auc, name)]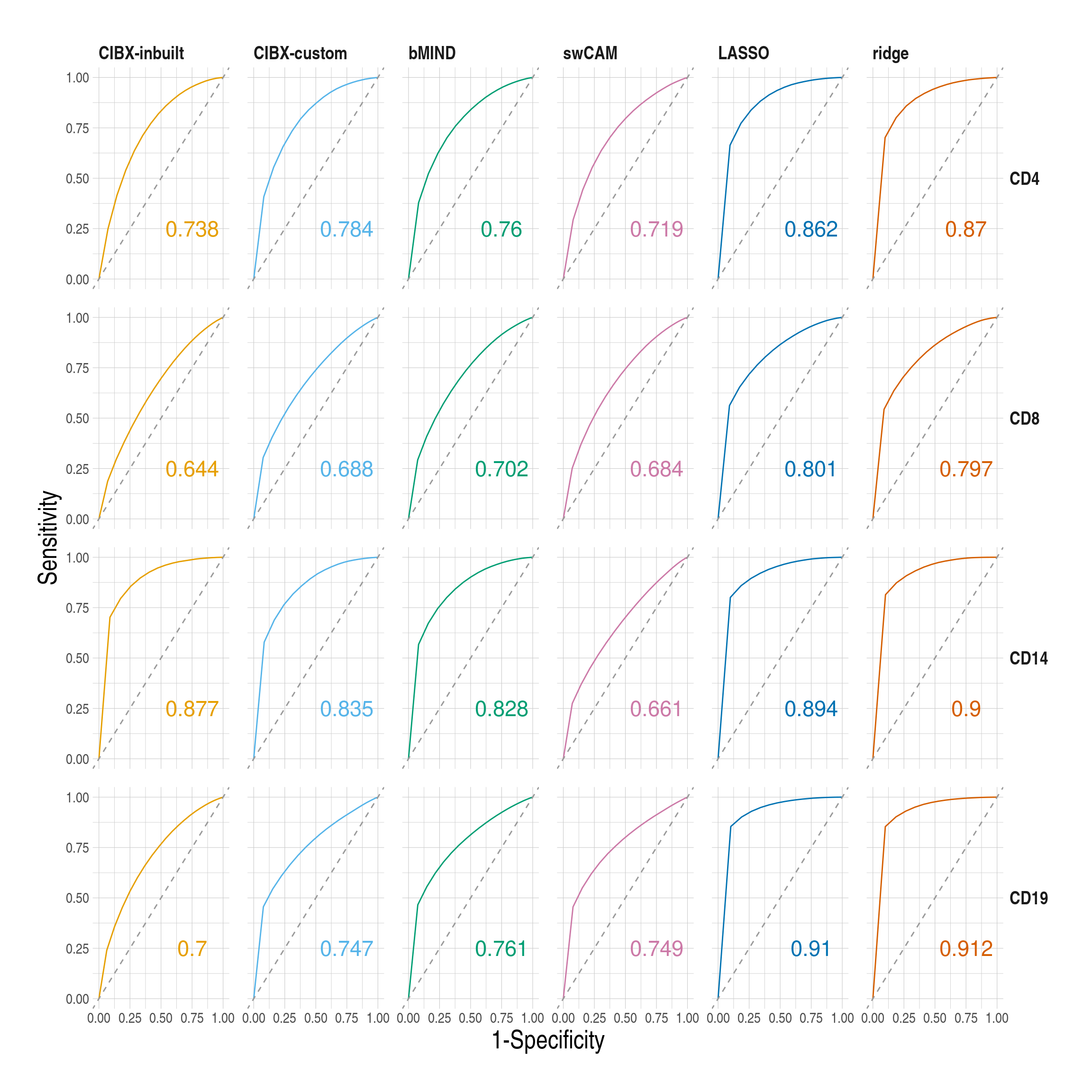
2.5 SupFig6, Train + Test samples, CLUSTER data
2.5.1 Common gene sets
Code
ggboxplot(
data = mychk[scenario == "common", ],
x = "approach", y = "auc", ylab = "AUC",
palette = appro.cols,
alpha = 0.35, fill = "approach",
add = c("jitter"),
add.params = list(shape = 21, size = 2.5, alpha = 1)
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(pheno ~ celltype, scales = "free") +
labs(fill = NULL) +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "top",
strip.text.y.right = element_text(angle = 0),
legend.text = element_text(size = rel(1))
) +
rremove("xlab")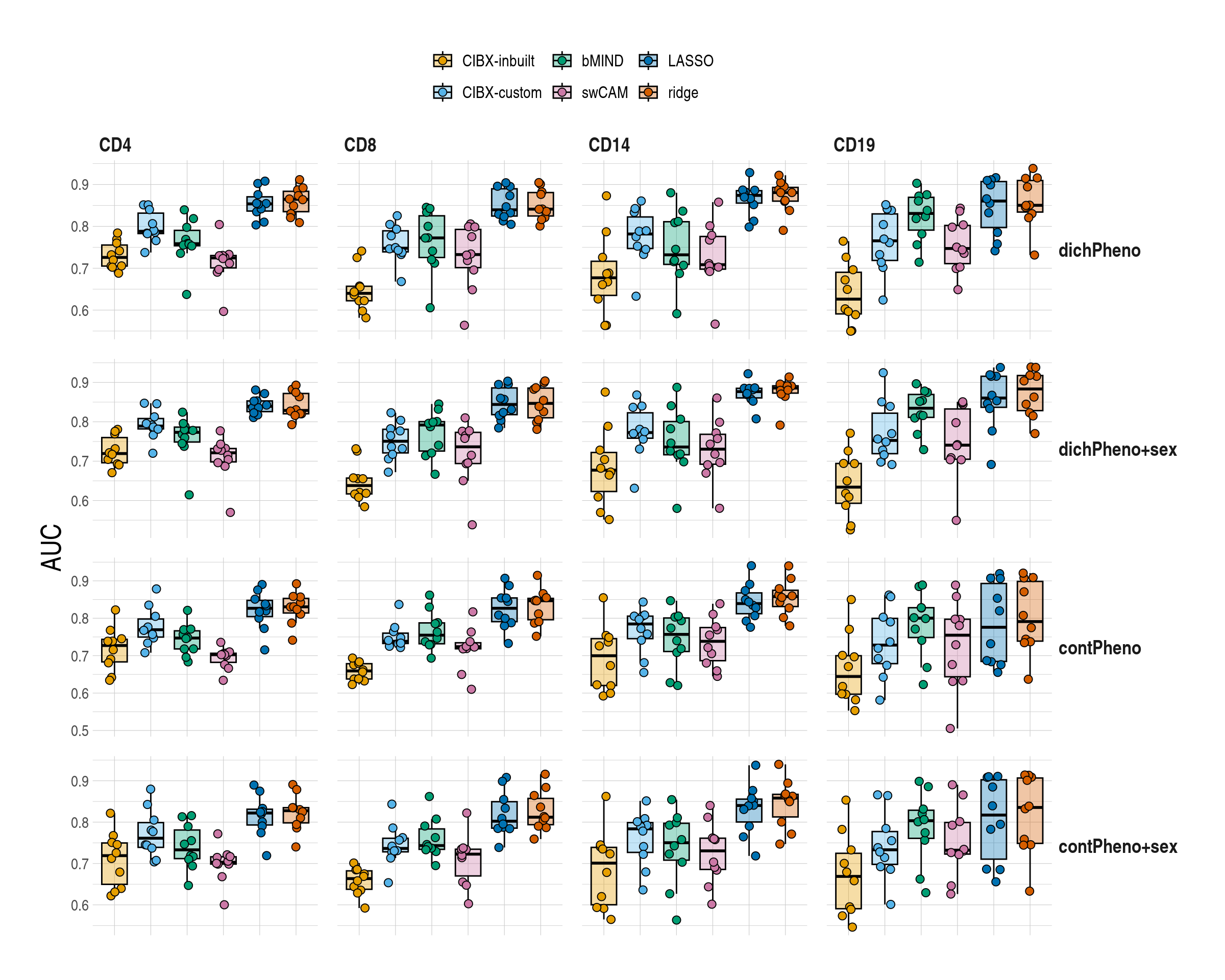
Code
tbl_out <- tbl_res[rows.order, c("celltype", grep("common", cols.order, value = TRUE))]
names(tbl_out) <- gsub("main_|common_", "", names(tbl_out))
kbl(
x = tbl_out, row.names = FALSE,
caption = "Median AUC (Q25-Q75) across 10 simulated phentoypes per cell by apparoch. **Genes common across approaches per cell type**"
) %>%
kable_paper("striped", full_width = FALSE) %>%
column_spec(column = 6:7, bold = T) %>%
pack_rows("dichotomous pheno", 1, 4,
label_row_css = "text-align: left;"
) %>%
pack_rows("dichotomous pheno + sex", 5, 8) %>%
pack_rows("continous pheno", 9, 12) %>%
pack_rows("continous pheno + sex", 13, 16)| celltype | inbuilt | custom | bMIND | swCAM | LASSO | RIDGE |
|---|---|---|---|---|---|---|
| dichotomous pheno | ||||||
| CD4 | 0.73 (0.71-0.76) | 0.79 (0.78-0.83) | 0.76 (0.75-0.79) | 0.72 (0.7-0.73) | 0.85 (0.84-0.87) | 0.86 (0.84-0.88) |
| CD8 | 0.64 (0.62-0.66) | 0.75 (0.74-0.79) | 0.77 (0.73-0.83) | 0.73 (0.7-0.79) | 0.84 (0.82-0.89) | 0.84 (0.82-0.88) |
| CD14 | 0.68 (0.64-0.72) | 0.78 (0.75-0.82) | 0.73 (0.71-0.81) | 0.71 (0.7-0.78) | 0.87 (0.86-0.89) | 0.88 (0.86-0.89) |
| CD19 | 0.63 (0.59-0.69) | 0.77 (0.72-0.83) | 0.83 (0.79-0.87) | 0.75 (0.71-0.8) | 0.86 (0.8-0.91) | 0.85 (0.83-0.91) |
| dichotomous pheno + sex | ||||||
| CD4 | 0.72 (0.7-0.76) | 0.79 (0.78-0.81) | 0.77 (0.75-0.79) | 0.72 (0.7-0.73) | 0.84 (0.83-0.85) | 0.83 (0.82-0.87) |
| CD8 | 0.64 (0.62-0.66) | 0.75 (0.72-0.78) | 0.79 (0.73-0.8) | 0.74 (0.69-0.77) | 0.84 (0.82-0.89) | 0.85 (0.81-0.89) |
| CD14 | 0.68 (0.62-0.72) | 0.77 (0.76-0.82) | 0.74 (0.72-0.8) | 0.73 (0.69-0.77) | 0.88 (0.86-0.89) | 0.89 (0.87-0.89) |
| CD19 | 0.63 (0.59-0.69) | 0.75 (0.72-0.82) | 0.83 (0.81-0.87) | 0.74 (0.7-0.83) | 0.86 (0.84-0.92) | 0.88 (0.83-0.92) |
| continous pheno | ||||||
| CD4 | 0.73 (0.68-0.74) | 0.77 (0.75-0.8) | 0.75 (0.72-0.77) | 0.7 (0.68-0.71) | 0.83 (0.8-0.85) | 0.83 (0.81-0.85) |
| CD8 | 0.66 (0.64-0.68) | 0.74 (0.73-0.76) | 0.75 (0.73-0.79) | 0.72 (0.72-0.73) | 0.83 (0.79-0.85) | 0.85 (0.8-0.85) |
| CD14 | 0.7 (0.62-0.75) | 0.78 (0.75-0.81) | 0.76 (0.71-0.8) | 0.74 (0.69-0.78) | 0.84 (0.81-0.87) | 0.86 (0.83-0.87) |
| CD19 | 0.64 (0.6-0.7) | 0.73 (0.68-0.8) | 0.8 (0.75-0.83) | 0.75 (0.64-0.8) | 0.78 (0.68-0.89) | 0.79 (0.74-0.9) |
| continous pheno + sex | ||||||
| CD4 | 0.72 (0.65-0.75) | 0.76 (0.74-0.8) | 0.73 (0.71-0.78) | 0.7 (0.7-0.72) | 0.82 (0.79-0.83) | 0.83 (0.8-0.83) |
| CD8 | 0.66 (0.64-0.68) | 0.74 (0.73-0.76) | 0.74 (0.73-0.78) | 0.72 (0.67-0.73) | 0.8 (0.79-0.85) | 0.81 (0.79-0.86) |
| CD14 | 0.7 (0.6-0.74) | 0.78 (0.73-0.8) | 0.75 (0.71-0.8) | 0.73 (0.69-0.76) | 0.84 (0.8-0.86) | 0.86 (0.81-0.87) |
| CD19 | 0.67 (0.59-0.72) | 0.73 (0.7-0.78) | 0.8 (0.76-0.83) | 0.73 (0.72-0.8) | 0.82 (0.71-0.9) | 0.84 (0.75-0.91) |
2.6 SupFig7, slopes
- Distributions of sensitivity (y-axis) & specificity (y-axis) by cell type and approach. An FDR < 0.05 were used in both imputed and observed data. Each point is a simulated phenotype.
Code
## sen and spe across phenotypes
mychks <- copy(annotdsn1)
mychks[, Sens := N_recSIG / N_SIG_obs][
, Spes := N_recNS / N_NS_obs
]
specresplt <- ggboxplot(
data = mychks, x = "approach",
y = "Spes", ylab = "Specificity",
palette = appro.cols,
alpha = 0.35, fill = "approach",
add.params = list(shape = 21, size = 3, alpha = 0.8),
add = c("jitter")
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(~celltype, scales = "free") +
used_ggthemes() + rremove("xlab") +
theme(
axis.text.x = element_blank(),
legend.text = element_text(size = rel(1.2))
) +
labs(fill = NULL)
sensresplt <- ggboxplot(
data = mychks, x = "approach",
y = "Sens", ylab = "Sensitivity",
palette = appro.cols,
alpha = 0.35, fill = "approach",
add.params = list(shape = 21, size = 3, alpha = 0.8),
add = c("jitter")
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(~celltype, scales = "free") +
used_ggthemes(
axis.text.x = element_blank(),
legend.text = element_text(size = rel(1.2))
) +
rremove("xlab") + labs(fill = NULL)
ggarrange(sensresplt, specresplt, nrow = 2, common.legend = TRUE)
figS7a <- copy(mychks)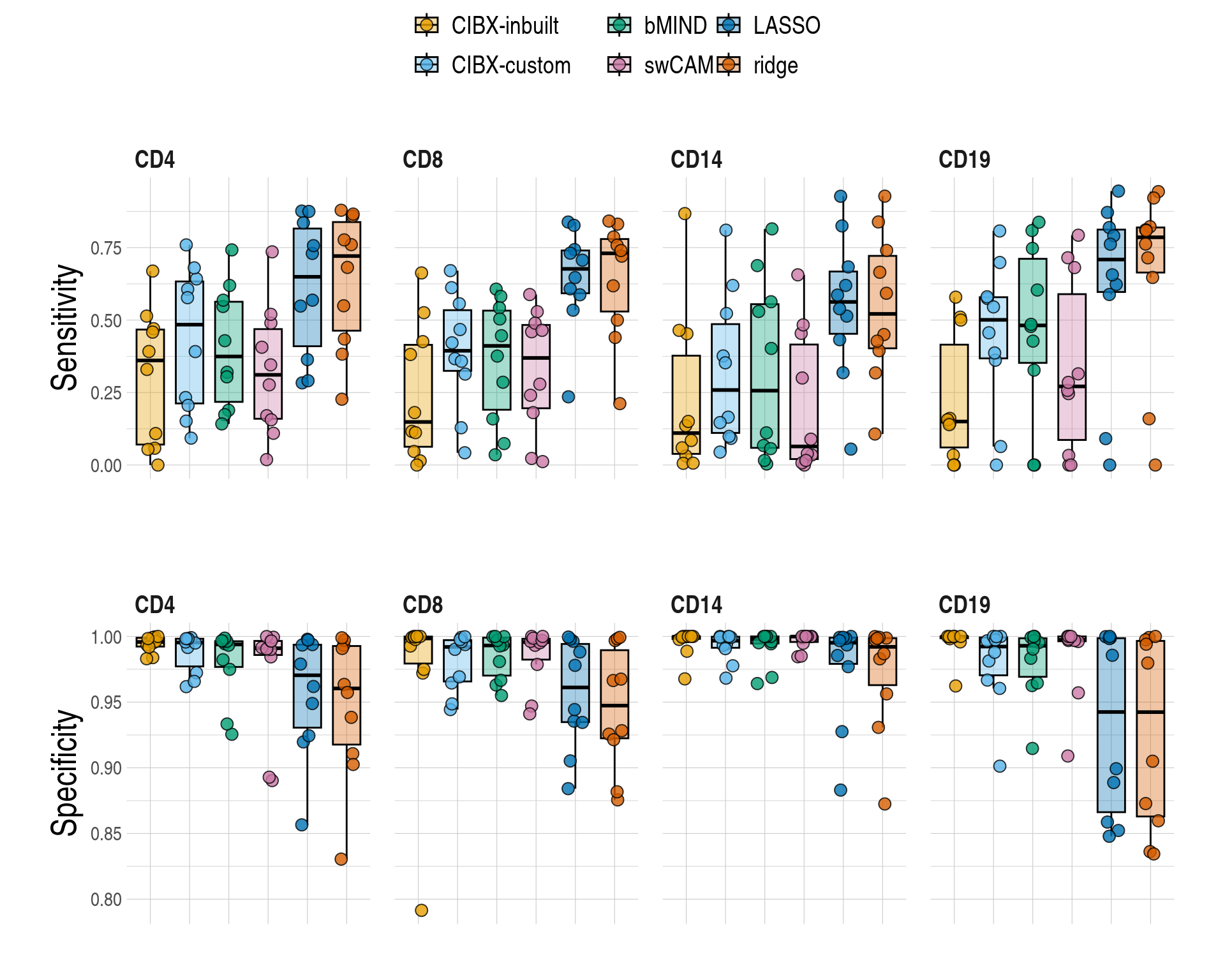
- Effect sizes (logFC), standard errors (S.E.) of effect sizes, z scores of effect size between observed (x-axis) and imputed (y-axis) data based on CD4 with one simulated phenotype
Code
y <- copy(degres)
head(y)
logFC AveExpr t P.Value adj.P.Val B s2post
1: 0.3554621 7.260532 8.525827 1.339332e-14 8.385557e-11 22.64075 0.06458024
2: 0.3090823 5.954265 8.394649 2.884120e-14 9.028738e-11 21.90352 0.05036503
3: 0.2993696 5.147788 8.049545 2.126368e-13 4.437730e-10 19.98378 0.05138763
4: 0.3058392 4.130806 7.938247 4.023035e-13 6.297056e-10 19.37123 0.05514713
5: 0.2602279 4.532937 7.783485 9.707277e-13 1.078018e-09 18.52517 0.04152844
6: 0.3030935 5.021438 7.772500 1.033079e-12 1.078018e-09 18.46538 0.05649601
Nsample Ncase Ncon impvt impvlogFC impvp impvadjp impvNsample
1: 151 66 85 5.287726 0.2487747 4.205486e-07 0.0002633055 151
2: 151 66 85 5.197207 0.2747704 6.374286e-07 0.0003325784 151
3: 151 66 85 4.384273 0.3168297 2.153044e-05 0.0018722509 151
4: 151 66 85 5.493173 0.3488366 1.609410e-07 0.0001439502 151
5: 151 66 85 4.416395 0.2156537 1.888024e-05 0.0017860222 151
6: 151 66 85 4.447224 0.3540465 1.663371e-05 0.0017506914 151
impvNcase impvNcon approach celltype fakedpheno gene truecol callmade
1: 66 85 inbuilt CD4 PC1 CD44 1Yes 1Yes
2: 66 85 inbuilt CD4 PC1 C1orf43 1Yes 1Yes
3: 66 85 inbuilt CD4 PC1 VCP 1Yes 1Yes
4: 66 85 inbuilt CD4 PC1 P2RX4 1Yes 1Yes
5: 66 85 inbuilt CD4 PC1 NAA20 1Yes 1Yes
6: 66 85 inbuilt CD4 PC1 PSME3 1Yes 1Yes
recovery
1: Yes-Sig
2: Yes-Sig
3: Yes-Sig
4: Yes-Sig
5: Yes-Sig
6: Yes-Sig
xx <- y[fakedpheno == "PC1" & celltype == "CD4"]
library(ggplot2)
library(cowplot)
theme_set(theme_cowplot())
xx[, class := paste(truecol, callmade, sep = "/")]
xx[, se := logFC / t]
xx[, impvse := impvlogFC / impvt]
xsum <- xx[, .(n = .N, medse = median(se), medimpvse = median(impvse)), by = c("class", "approach")]
xsum[, p := n / sum(n), by = "approach"]
xsum[order(approach, class)]
class approach n medse medimpvse p
1: 0No/0Nonsig inbuilt 4187 0.05508787 0.05873184 0.668743012
2: 0No/1Yes inbuilt 27 0.06178480 0.05758701 0.004312410
3: 1Yes/0Nonsig inbuilt 1372 0.05195210 0.05968275 0.219134324
4: 1Yes/1Yes inbuilt 675 0.05002479 0.05390566 0.107810254
5: 0No/0Nonsig custom 4294 0.05983496 0.05406578 0.593012015
6: 0No/1Yes custom 122 0.06114711 0.05215159 0.016848502
7: 1Yes/0Nonsig custom 1011 0.05544873 0.05477584 0.139621599
8: 1Yes/1Yes custom 1814 0.05195309 0.04837343 0.250517884
9: 0No/0Nonsig bMIND 9844 0.05656370 0.07044463 0.521646972
10: 0No/1Yes bMIND 253 0.05715402 0.06521827 0.013406815
11: 1Yes/0Nonsig bMIND 3340 0.05657640 0.07587389 0.176991150
12: 1Yes/1Yes bMIND 5434 0.05345500 0.06754613 0.287955063
13: 0No/0Nonsig swCAM 9940 0.05655989 0.08271920 0.526734142
14: 0No/1Yes swCAM 157 0.05816015 0.07310080 0.008319644
15: 1Yes/0Nonsig swCAM 4477 0.05538832 0.10034547 0.237242330
16: 1Yes/1Yes swCAM 4297 0.05354609 0.09459480 0.227703884
17: 0No/0Nonsig LASSO 9286 0.05643993 0.04478155 0.492077791
18: 0No/1Yes LASSO 811 0.05817531 0.04490354 0.042975995
19: 1Yes/0Nonsig LASSO 1443 0.05524053 0.04429984 0.076466536
20: 1Yes/1Yes LASSO 7331 0.05416448 0.04387849 0.388479678
21: 0No/0Nonsig RIDGE 9196 0.05632371 0.04528211 0.487308569
22: 0No/1Yes RIDGE 901 0.05933682 0.04601117 0.047745218
23: 1Yes/0Nonsig RIDGE 1240 0.05582991 0.04509011 0.065709289
24: 1Yes/1Yes RIDGE 7534 0.05411614 0.04438538 0.399236924
class approach n medse medimpvse pDistributions of r-squared (Rsq, y-axis) of imputed log\(_{2}\) fold changes (FC) regression on observed effect sizes by approach.
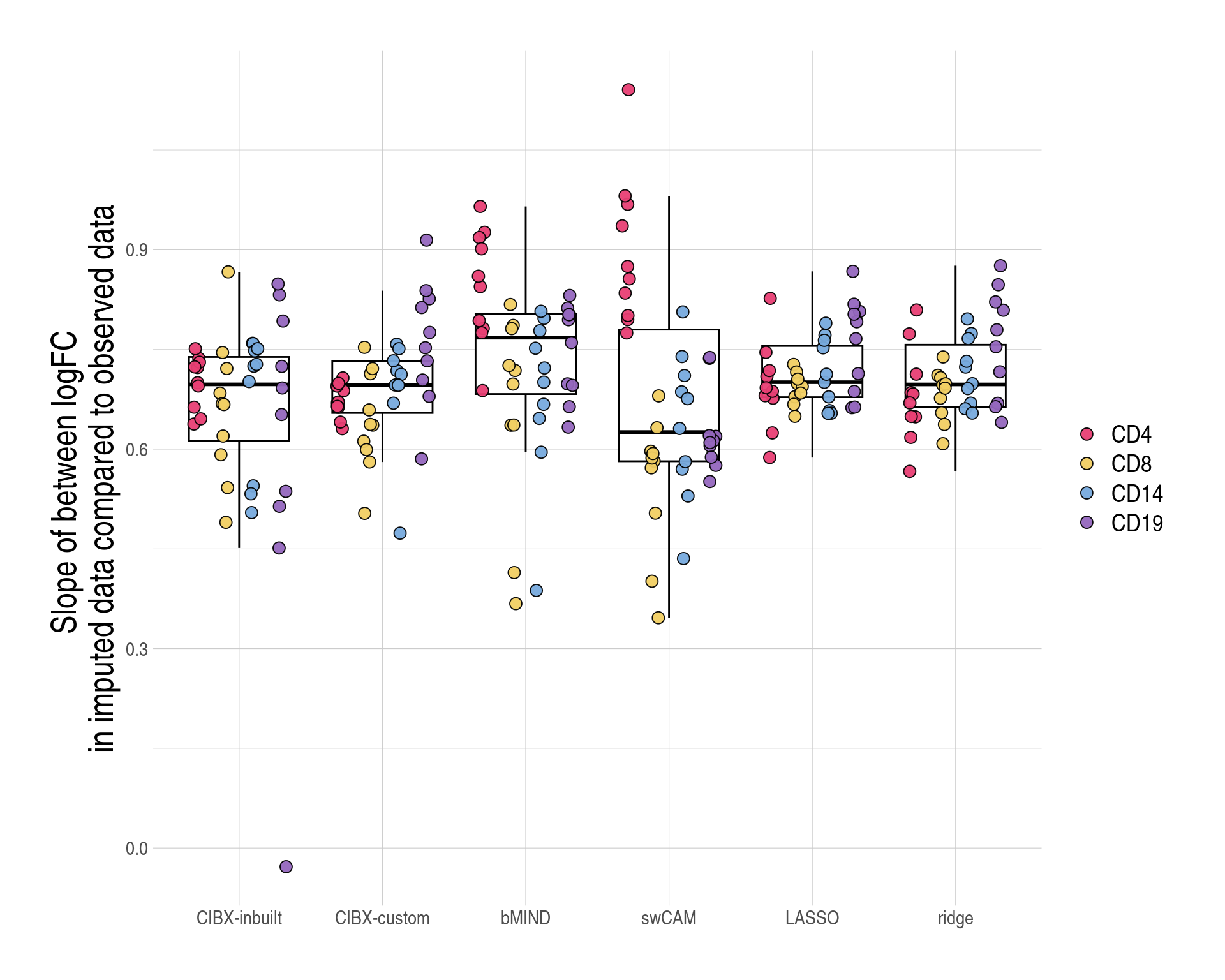
Distributions of slopes (y-axis) of imputed log\(_{2}\) fold changes (FC) regression on observed effect sizes by approach. Each point is a simulated phenotype, coloured by cell type.
Code
library(magrittr)
calc_rsq <- function(x, y) {
summ <- lm(y ~ x) %>% summary()
summ$r.squared
}
calc_slope <- function(x, y) {
cf <- lm(y ~ x - 1) %>% coef()
}
ysum <- y[, .(logFC_rsq = calc_rsq(logFC, impvlogFC), logFC_slope = calc_slope(logFC, impvlogFC)),
by = c("approach", "fakedpheno", "celltype")
]
## colors for cell type
ctfrat.pal <- friend11[seq(length(sctorder))]
## plot b
ysum[, approach := factor(approach,
levels = expr_order,
labels = appro_lab
)]
ggboxplot(
data = ysum, x = "approach",
y = "logFC_rsq",
palette = ctfrat.pal,
ylab = "Rsq between logFC\nin imputed and observed data",
add = "jitter",
add.params = list(
fill = "celltype",
alpha = 0.95, shape = 21,
size = 3
),
ggtheme = used_ggthemes(
legend.text = element_text(size = rel(1.2))
)
) +
labs(fill = NULL) + rremove("xlab")
## plot c
ggboxplot(
data = ysum, x = "approach",
y = "logFC_slope",
ylab = "Slope of between logFC \nin imputed data compared to observed data",
palette = ctfrat.pal,
add = "jitter",
add.params = list(fill = "celltype", alpha = 0.95, shape = 21, size = 3),
ggtheme = used_ggthemes(legend.text = element_text(size = rel(1.2)))
) +
labs(fill = NULL) +
rremove("xlab")
figS7bc <- copy(ysum)
## mean logFC slope across simulated phentoypes by approach and cell type
ysumres <- ysum[,
{
xx <- round(mean(logFC_slope), 2)
},
by = c("approach", "celltype")
]
## mean logFC slop across cell types by approach
ysumres[,
{
xxmin <- range(V1)[1]
xxmax <- range(V1)[2]
list(xxmin, xxmax)
},
by = "approach"
]
approach xxmin xxmax
1: CIBX-inbuilt 0.60 0.70
2: CIBX-custom 0.64 0.76
3: bMIND 0.66 0.85
4: swCAM 0.55 0.90
5: LASSO 0.69 0.76
6: ridge 0.68 0.76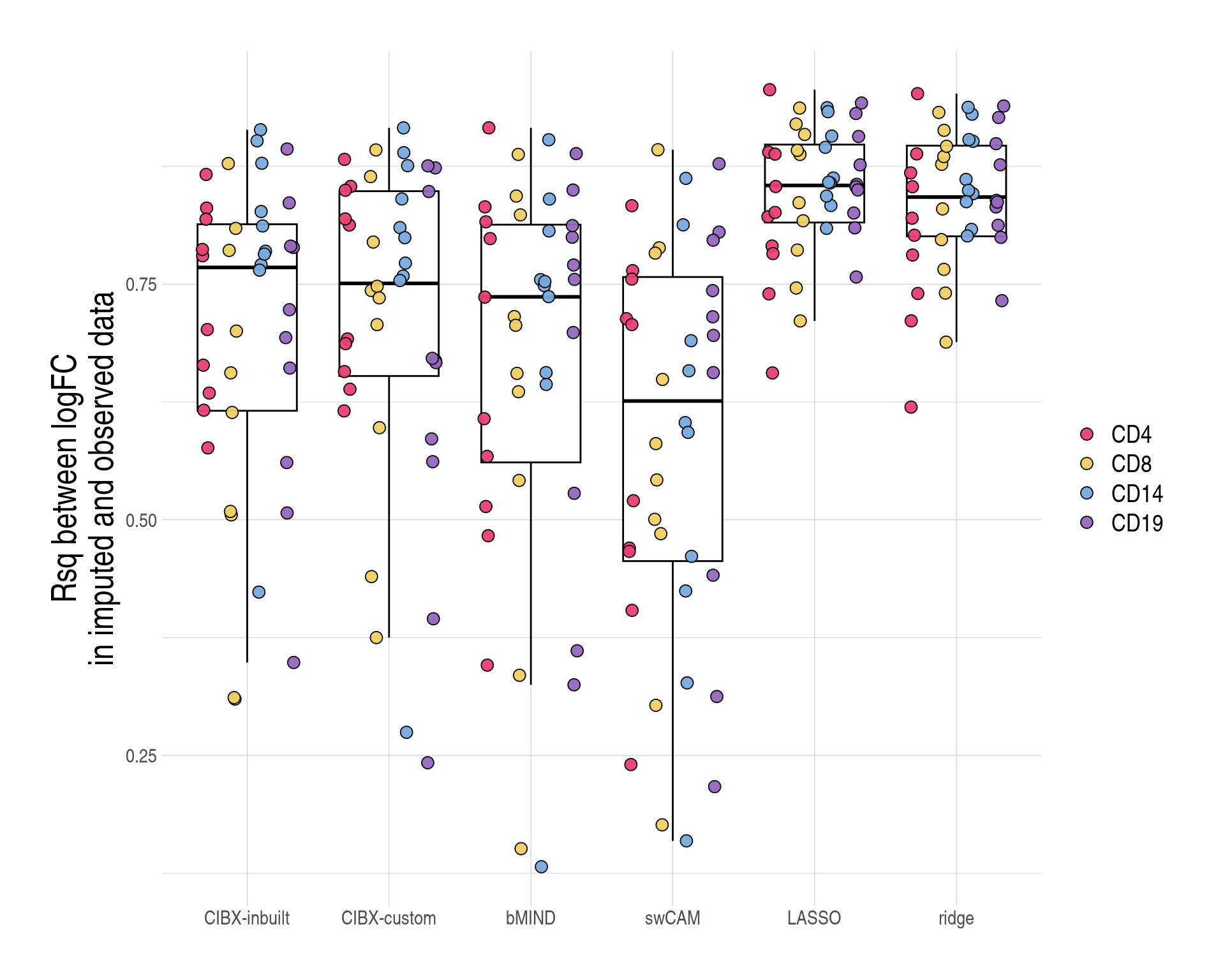
2.7 SupFig8, AUCs in pseudo datasets based on common gene sets
Code
simres_pltlist$common$sce1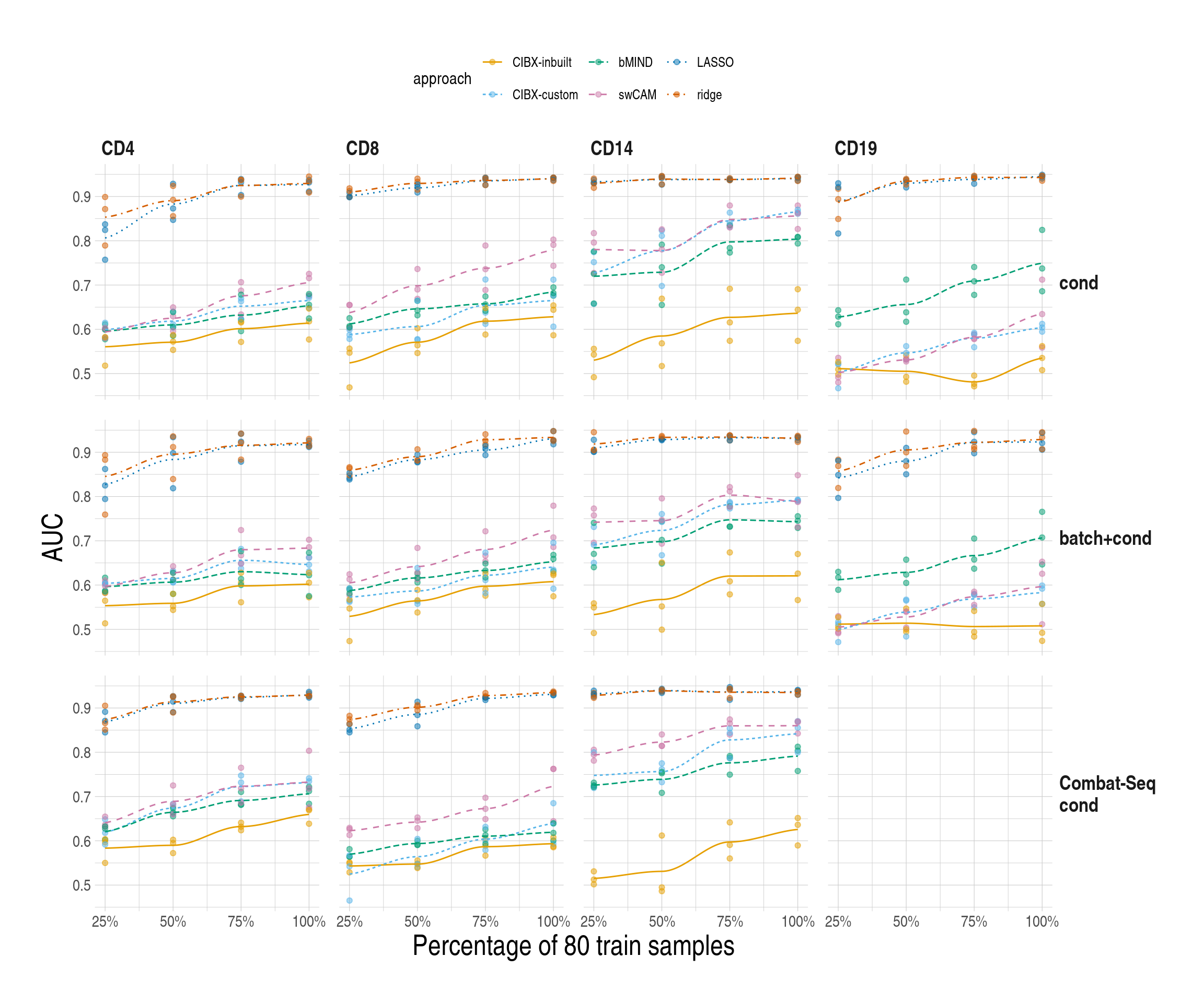
2.8 SupFig9, SupTab2, SupTab3
Code
## cluster data
proj_dir <- here::here()
proj_dir
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen"
aa_files <- list.files(file.path(proj_dir, "CLUSTERdeconRes/workflowInfo"),
"pipeline_trace*",
full.names = TRUE
)
aadata <- lapply(aa_files, fread) %>% set_names(., basename(aa_files))
lapply(aadata, nrow)
$`pipeline_trace_2024-10-12_23-09-13.txt`
[1] 2
$`pipeline_trace_2024-10-12_23-34-29.txt`
[1] 622
$`pipeline_trace_2024-10-16_18-31-30.txt`
[1] 13
$`pipeline_trace_2024-10-16_18-39-21.txt`
[1] 5
## pipeline_trace_2024-10-12_23-34-29.txt: full run, but CIBX ran on 4 cpus
## pipeline_trace_2024-10-16_18-39-21.txt; rerun CIBX with 8 cpus
nextflowlog <- aadata[["pipeline_trace_2024-10-12_23-34-29.txt"]][!(name %in% aadata[["pipeline_trace_2024-10-16_18-39-21.txt"]]$name), ] %>%
rbind(aadata[["pipeline_trace_2024-10-16_18-39-21.txt"]], .)
nextflowlog[, .N] # 622
[1] 622
nextflowlog <- nextflowlog[status != "FAILED", ]
nextflowlog[, .N] # 622
[1] 622
## check if realtime ==0
stopifnot(all(vtimesec(nextflowlog$realtime) != 0))
## memory units in the log
stringr::str_extract_all(nextflowlog$vmem, "[[:alpha:]]+$") |>
unique() |>
unlist()
[1] "MB" "GB"
## check if memory format not captured by default
nextflowlog[!stringr::str_detect(nextflowlog$vmem, "[[:alpha:]]+$")]
Empty data.table (0 rows and 17 cols): task_id,name,tag,status,exit,realtime...
stopifnot(all(!is.na(vmem.usage(nextflowlog$vmem))))
## unique process
uni_procs <- stringr::str_extract(nextflowlog$name, "[[:alnum:]]+") |> unique()
uni_procs
[1] "ZENODO" "PREPCLUSTER" "cibxrun" "runSWCAM" "runBMIND"
[6] "runPenalised"
nextflowlog[grepl(paste0(uni_procs[1:2], collapse = "|"), name), .N] # 2
[1] 2
## exclude data prep steps
nextflowlog <- nextflowlog[!(tag %in% c("zenodo", "prepcluster")), ]
nextflowlog$trainper <- 100
nextflowlog$simdataid <- "cluster"
nextflowlog[grep("^cibxrun:LM22DECONVRUN", name), approach := "inbuilt"]
nextflowlog[grep("^cibxrun:DECONVRUN[[:space:]]", name), approach := "custom"]
nextflowlog[grep("^runBMIND:BMIND[[:space:]]", name), approach := "bMIND"]
nextflowlog[grep("^runSWCAM:SWCAM", name), approach := "swCAM"]
nextflowlog[grep("^runPenalised:PREPCHUNK", name), approach := "chunkprep"]
nextflowlog[grepl("^runPenalised", name) & grepl("lasso", tag), approach := "LASSO"]
nextflowlog[grepl("^runPenalised", name) & grepl("ridge", tag), approach := "RIDGE"]
nextflowlog[, table(approach)]
approach
bMIND chunkprep custom inbuilt LASSO RIDGE swCAM
1 4 1 1 304 304 2
## na for fraction deconvolution or gene signature
nextflowlog[is.na(approach), name]
[1] "cibxrun:DECONVRUNGS (CIBXgs_cluster)"
[2] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_cluster)"
[3] "runBMIND:BMINDFRACT (bmindrunfract_cluster)"
expr_order %in% nextflowlog$approach
[1] TRUE TRUE TRUE TRUE TRUE TRUE
rundata4plt <- lapply(expr_order, function(appin) {
nextflowlog[approach == appin, ][,
{
ncpu <- unique(cpus) %>% paste0(., collapse = "/")
cputimes <- sum(vtimesec(realtime) / 60) %>% round(., 2)
memt <- median(vmem.usage(vmem) / 1024) %>% round(., 2)
list(ncpu, cputimes, memt)
},
by = c("simdataid", "trainper")
]
}) %>%
set_names(., expr_order) %>%
rbindlist(., idcol = "approach")
## per chunk cpu time & memory for LASSO and ridge
expr_order[5:6]
[1] "LASSO" "RIDGE"
cluster_perchunk <- lapply(expr_order[5:6], function(appin) {
nextflowlog[approach == appin, ][,
{
ncpu <- unique(cpus) %>% paste0(., collapse = "/")
cputimes <- vtimesec(realtime) / 60
memt <- vmem.usage(vmem) / 1024
nchunk <- .N
Q50c <- median(cputimes) %>% round(., 2)
Q2575c <- quantile(cputimes, c(0.25, 0.75)) %>%
round(., 2) %>%
paste0(., collapse = ",")
Q50m <- median(memt) %>% round(., 2)
Q2575m <- quantile(memt, c(0.25, 0.75)) %>%
round(., 2) %>%
paste0(., collapse = ",")
list(ncpu,
nchunk,
cputimes_pchunk = paste0(Q50c, " (", Q2575c, ")"),
memt_pchunk = paste0(Q50m, " (", Q2575m, ")")
)
},
by = c("simdataid", "trainper")
]
}) %>%
set_names(., expr_order[5:6]) %>%
rbindlist(., idcol = "approach")
rundata4plt[cluster_perchunk, Nchunks := i.nchunk, on = "approach"]
rundata4plt[cluster_perchunk, ":="(
cpu_perchunk = i.cputimes_pchunk,
mem_perchunk = i.memt_pchunk
), on = "approach"]
rundata4plt[, trainper := as.numeric(trainper)]
rundata4plt[, approach := appro_lab[match(rundata4plt$approach, names(appro_lab))]]
rundata4plt[, approach := factor(approach, appro_lab)]
appro.cols.mod <- appro.cols
names(appro.cols.mod) <- appro_lab
##
## cpu time
cluster_cputime <- rundata4plt[, tstrsplit(cputimes, " ", keep = 1L) %>% unlist() %>% as.numeric()]
cluster_cputime
[1] 8.80 4.45 12.00 1713.13 624.22 2928.45
## relative to CIBX
(cluster_cputime / cluster_cputime[2]) %>% round(., 2)
[1] 1.98 1.00 2.70 384.97 140.27 658.08
## mem
cluster_mem <- rundata4plt[, tstrsplit(memt, " ", keep = 1L) %>% unlist() %>% as.numeric()]
cluster_mem
[1] 11.00 5.50 1.80 2.95 16.70 58.50
## relative to CIBX
(cluster_mem / cluster_mem[2]) %>% round(., 2)
[1] 2.00 1.00 0.33 0.54 3.04 10.64Code
setdiff(names(rundata4plt), c("simdataid", "trainper")) %>%
rundata4plt[, ., with = FALSE] %>%
kable(., caption = "resource usage metrics for the CLUSTER data")| approach | ncpu | cputimes | memt | Nchunks | cpu_perchunk | mem_perchunk |
|---|---|---|---|---|---|---|
| CIBX-inbuilt | 8 | 8.80 | 11.00 | NA | NA | NA |
| CIBX-custom | 8 | 4.45 | 5.50 | NA | NA | NA |
| bMIND | 4 | 12.00 | 1.80 | NA | NA | NA |
| swCAM | 10/1 | 1713.13 | 2.95 | NA | NA | NA |
| LASSO | 1 | 624.22 | 16.70 | 304 | 1.84 (1.23,2.73) | 16.7 (11.7,24.8) |
| ridge | 1 | 2928.45 | 58.50 | 304 | 8.85 (6.09,12.78) | 58.5 (39.18,81.22) |
2.9 resource usage metrics for simdata
- 3 simulation data (N=160 samples) x Combat-seq (Yes/No) x training percentage (25, 50, 75, 100)
Code
## simData
proj_dir <- here::here()
proj_dir
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen"
## runlog
nextflowlog <- file.path(
proj_dir,
"simDataRes/workflowInfo/pipeline_trace_2024-10-12_22-15-21.txt"
) %>% fread()
nextflowlog[, .N] # 11073
[1] 11073
nextflowlog <- nextflowlog[status != "FAILED", ]
nextflowlog[, .N] # 11073
[1] 11073
## check if realtime ==0
stopifnot(all(vtimesec(nextflowlog$realtime) != 0))
## memory units in the log
stringr::str_extract_all(nextflowlog$vmem, "[[:alpha:]]+$") |>
unique() |>
unlist()
[1] "GB" "MB"
## check if memory format not captured by default
nextflowlog[!stringr::str_detect(nextflowlog$vmem, "[[:alpha:]]+$")]
Empty data.table (0 rows and 17 cols): task_id,name,tag,status,exit,realtime...
stopifnot(all(!is.na(vmem.usage(nextflowlog$vmem))))
## unique process
uni_procs <- stringr::str_extract(nextflowlog$name, "[[:alnum:]]+") |> unique()
uni_procs
[1] "PREPSCEQTL" "CBATSIMDATA" "SIMDATA" "SIMDECONVINPUT"
[5] "runSWCAM" "cibxrun" "runBMIND" "runPenalised"
nextflowlog[grepl(paste0(uni_procs[1:4], collapse = "|"), name), .N] # 31
[1] 31
## tag="-" for data prep steps
nextflowlog[tag == "-", name] %>% length()
[1] 31
## exclude data prep steps
nextflowlog <- nextflowlog[tag != "-", ]
nextflowlog$trainper <- str_extract(nextflowlog$tag, "[[:digit:]]+$")
nextflowlog$simdataid <- str_extract(nextflowlog$tag, "1Msim.+$")
stopifnot(all(!is.na(nextflowlog$simdataid)))
nextflowlog[grep("^cibxrun:LM22DECONVRUN", name), approach := "inbuilt"]
nextflowlog[grep("^cibxrun:DECONVRUN[[:space:]]", name), approach := "custom"]
nextflowlog[grep("^runBMIND:BMIND[[:space:]]", name), approach := "bMIND"]
nextflowlog[grep("^runSWCAM:SWCAM", name), approach := "swCAM"]
nextflowlog[grep("^runPenalised:PREPCHUNK", name), approach := "chunkprep"]
nextflowlog[grepl("^runPenalised", name) & grepl("lasso", tag), approach := "LASSO"]
nextflowlog[grepl("^runPenalised", name) & grepl("ridge", tag), approach := "RIDGE"]
nextflowlog[, table(approach)]
approach
bMIND chunkprep custom inbuilt LASSO RIDGE swCAM
24 96 24 24 5377 5377 48
## na for fraction deconvolution or gene signature
nextflowlog[is.na(approach), name]
[1] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.2.50)"
[2] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.1.75)"
[3] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.3.100)"
[4] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.1.75)"
[5] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.3.50)"
[6] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.2.75)"
[7] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.2.100)"
[8] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.2.25)"
[9] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.3.100)"
[10] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.3.25)"
[11] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.2.50)"
[12] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.1.25)"
[13] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.2.50)"
[14] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.1.50)"
[15] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.3.25)"
[16] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.1.25)"
[17] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.3.50)"
[18] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.2.25)"
[19] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.3.75)"
[20] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.3.100)"
[21] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.1.100)"
[22] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.3.50)"
[23] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.1.100)"
[24] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.2.75)"
[25] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.2.50)"
[26] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.1.100)"
[27] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.2.25)"
[28] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.3.50)"
[29] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.1.75)"
[30] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.1.50)"
[31] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.3.25)"
[32] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.2.100)"
[33] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.3.100)"
[34] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_dat.160.2.100)"
[35] "runSWCAM:DEBCAMFRACTS80 (debCAMfracts80_1Msim_Combatdat.160.3.75)"
[36] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.2.75)"
[37] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.1.100)"
[38] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.1.25)"
[39] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.2.25)"
[40] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.3.25)"
[41] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.2.50)"
[42] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.1.75)"
[43] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.2.100)"
[44] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.3.100)"
[45] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.1.25)"
[46] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.3.25)"
[47] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.2.75)"
[48] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.2.25)"
[49] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.3.25)"
[50] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.2.50)"
[51] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.3.50)"
[52] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.1.50)"
[53] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.1.25)"
[54] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.2.75)"
[55] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.1.75)"
[56] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.2.25)"
[57] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.1.50)"
[58] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.3.100)"
[59] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.3.50)"
[60] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.1.75)"
[61] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.2.75)"
[62] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.1.50)"
[63] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.2.100)"
[64] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.1.50)"
[65] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.2.100)"
[66] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.1.100)"
[67] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.1.100)"
[68] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_dat.160.3.75)"
[69] "cibxrun:DECONVRUNGS (CIBXgs_1Msim_Combatdat.160.3.75)"
[70] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.3.75)"
[71] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_dat.160.1.25)"
[72] "runBMIND:BMINDFRACT (bmindrunfract_1Msim_Combatdat.160.3.75)"
expr_order %in% nextflowlog$approach
[1] TRUE TRUE TRUE TRUE TRUE TRUE
rundata4plt <- lapply(expr_order, function(appin) {
nextflowlog[approach == appin, ][,
{
ncpu <- unique(cpus) %>% paste0(., collapse = "/")
cputimes <- sum(vtimesec(realtime) / 60) %>% round(., 2)
memt <- median(vmem.usage(vmem) / 1024) %>% round(., 2)
list(ncpu, cputimes, memt)
},
by = c("simdataid", "trainper")
]
}) %>%
set_names(., expr_order) %>%
rbindlist(., idcol = "approach")
rundata4plt[, trainper := as.numeric(trainper)]
rundata4plt[, approach := appro_lab[match(rundata4plt$approach, names(appro_lab))]]
rundata4plt[, approach := factor(approach, appro_lab)]
appro.cols.mod <- appro.cols
names(appro.cols.mod) <- appro_labCode
figS9 <- rundata4plt
re_ws <- c("cputimes", "memt")
lapply(re_ws, function(resin) {
yyl <- ifelse(resin == "cputimes",
"log(CPU realtime in mins)",
"log(memory GB usage)"
)
ggplot(
data = rundata4plt,
aes(
x = trainper, y = log(!!sym(resin)),
colour = approach
)
) +
geom_point(alpha = 0.5, size = 1.5) +
geom_smooth(
aes(
group = approach,
colour = approach,
linetype = approach,
fill = approach
),
alpha = 0.35, se = FALSE, linewidth = 0.5
) +
scale_colour_manual(values = appro.cols.mod) +
scale_fill_manual(values = appro.cols.mod) +
scale_x_continuous(
breaks = seq(25, 100, by = 25),
labels = scales::label_percent(scale = 1)
) +
ylab(yyl) +
xlab("Percentage of 80 train samples") +
used_ggthemes(
legend.position = "top"
)
}) %>%
ggarrange(
plotlist = ., common.legend = TRUE,
labels = LETTERS
)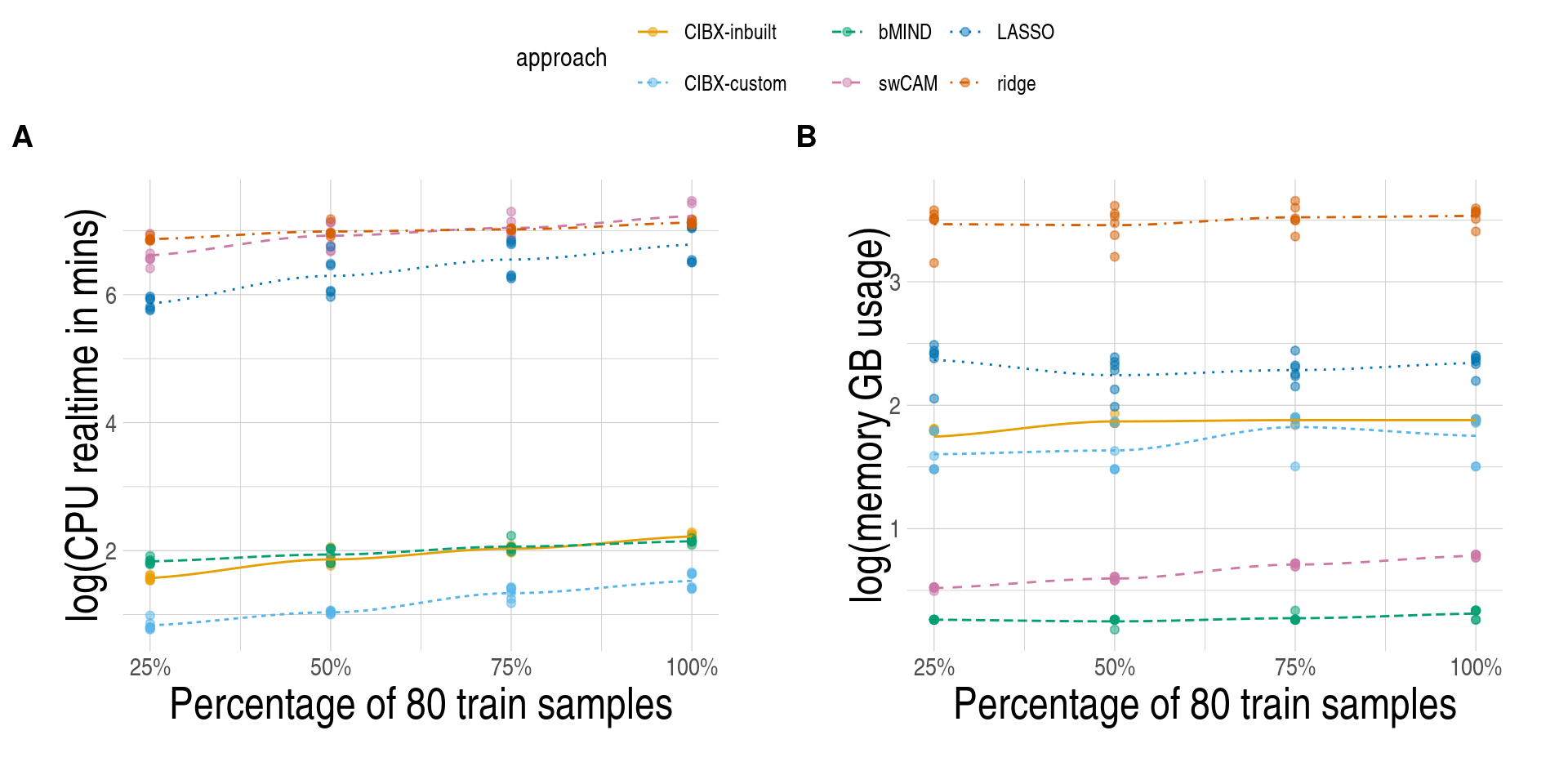
Code
## per chunk cpu time & memory for LASSO and ridge
## across 24 simulation data
expr_order[5:6]
[1] "LASSO" "RIDGE"
cluster_perchunk <- lapply(expr_order[5:6], function(appin) {
nextflowlog[approach == appin, ][
, {
ncpu <- unique(cpus) %>% paste0(., collapse = "/")
cputimes <- vtimesec(realtime) / 60
memt <- vmem.usage(vmem) / 1024
nchunk <- .N
Q50c <- median(cputimes) %>% round(., 2)
Q2575c <- quantile(cputimes, c(0.25, 0.75)) %>%
round(., 2) %>%
paste0(., collapse = ",")
Q50m <- median(memt) %>% round(., 2)
Q2575m <- quantile(memt, c(0.25, 0.75)) %>%
round(., 2) %>%
paste0(., collapse = ",")
list(ncpu,
nchunk,
cputimes_pchunk = paste0(Q50c, " (", Q2575c, ")"),
memt_pchunk = paste0(Q50m, " (", Q2575m, ")")
)
} # , by = c("simdataid", "trainper")
]
}) %>%
set_names(., expr_order[5:6]) %>%
rbindlist(., idcol = "approach")
cluster_perchunk[, approach := appro_lab[match(cluster_perchunk$approach, names(appro_lab))]]
cluster_perchunk[, approach := factor(approach, appro_lab)]
rundata_sim <- rundata4plt[,
{
ncpus <- unique(ncpu)
Nsimdata <- .N
Q50c <- median(cputimes) %>% round(., 2)
Q2575c <- quantile(cputimes, c(0.25, 0.75)) %>%
round(., 2) %>%
paste0(., collapse = ",")
Q50m <- median(memt) %>% round(., 2)
Q2575m <- quantile(memt, c(0.25, 0.75)) %>%
round(., 2) %>%
paste0(., collapse = ",")
list(ncpus,
Ndata = Nsimdata,
CPUmins = paste0(Q50c, " (", Q2575c, ")"),
memGB = paste0(Q50m, " (", Q2575m, ")")
)
},
by = "approach"
]
rundata_sim[cluster_perchunk, Nchunks := i.nchunk, on = "approach"]
rundata_sim[cluster_perchunk, ":="(
cpu_perchunk = i.cputimes_pchunk,
mem_perchunk = i.memt_pchunk
), on = "approach"]
## cpu time
sim_cputime <- rundata_sim[, tstrsplit(CPUmins, " ", keep = 1L) %>% unlist() %>% as.numeric()]
sim_cputime
[1] 7.22 3.08 7.61 1103.68 592.13 1103.60
## relative to CIBX
(sim_cputime / sim_cputime[2]) %>% round(., 2)
[1] 2.34 1.00 2.47 358.34 192.25 358.31
## mem
sim_mem <- rundata_sim[, tstrsplit(memGB, " ", keep = 1L) %>% unlist() %>% as.numeric()]
sim_mem
[1] 6.45 6.00 1.30 1.92 10.40 33.60
## relative to CIBX
(sim_mem / sim_mem[2]) %>% round(., 2)
[1] 1.07 1.00 0.22 0.32 1.73 5.60
kable(rundata_sim, caption = "resource usage metrics for the simulation data")| approach | ncpus | Ndata | CPUmins | memGB | Nchunks | cpu_perchunk | mem_perchunk |
|---|---|---|---|---|---|---|---|
| CIBX-inbuilt | 4 | 24 | 7.22 (5.64,8.12) | 6.45 (6.25,6.6) | NA | NA | NA |
| CIBX-custom | 4 | 24 | 3.08 (2.71,4.08) | 6 (4.47,6.53) | NA | NA | NA |
| bMIND | 4 | 24 | 7.61 (6.33,8.18) | 1.3 (1.3,1.3) | NA | NA | NA |
| swCAM | 10/1 | 24 | 1103.68 (799.36,1256.04) | 1.92 (1.77,2.07) | NA | NA | NA |
| LASSO | 1 | 24 | 592.13 (391.41,862.2) | 10.4 (9.46,10.94) | 5377 | 1.88 (0.54,3.85) | 10.2 (4.4,15.9) |
| ridge | 1 | 24 | 1103.6 (1017.01,1199.88) | 33.6 (32.83,35.35) | 5377 | 4.62 (2.1,7.25) | 33.1 (15.3,52.2) |
2.10 SupFig10, Inbuilt and custom signature Genes, and debCAM cell type-specific genes
Code
lm22mtx_f <- file.path("CIBXres/LM22deconv", runid) %>%
file.path(data_dir, .) %>%
file.path(., "sigMTX4dev.txt")
cumtx_f <- file.path("CIBXres/GS", runid) %>%
file.path(data_dir, .) %>%
file.path(., "CIBERSORTx_refexpr4CIBXgsClass.CIBERSORTx_refexpr4CIBXgs.bm.K999.txt")
## CIBX-inbuilt and custom signature genes
sigmtx_files <- c(lm22mtx_f, cumtx_f)
## signature matrcies, inbuilt and custom,
sigmtxs <- lapply(sigmtx_files, function(filein) {
datin <- fread(filein)
pltmat <- datin[, -1] %>% as.matrix()
rownames(pltmat) <- datin[[1]]
if (ncol(pltmat) != 4) {
ourorder <- lm22merged
names(ourorder) <- colnames(pltmat)
othercls <- setdiff(unique(ourorder), sctorder)
ourorder <- sort(factor(ourorder, levels = c(sctorder, othercls)))
pltmat <- pltmat[, names(ourorder)]
} else {
pltmat <- pltmat[, sctorder]
ourorder <- NULL
}
min_max <- pheatmap:::scale_mat(pltmat, "row") %>% range()
col_fun <- circlize::colorRamp2(
seq(from = min_max[1], to = min_max[2], length.out = 11),
cols4all::c4a("brewer.rd_bu", n = 11, reverse = TRUE)
)
list(
pltmat = pltmat,
col_fun = col_fun,
matorder = ourorder
)
})
names(sigmtxs) <- c("inbuilt", "custom")
figS10ab <- lapply(sigmtxs, "[[", "pltmat")
names(figS10ab) <- paste0("figS10", letters[seq_along(figS10ab)])
## Lm22 merged text
ourorder <- sigmtxs$inbuilt$matorder
descri.text <- NULL
for (cttype in unique(ourorder)) {
descri.text <- c(
descri.text,
sprintf(
"%s: %s",
cttype,
names(ourorder)[ourorder == cttype] %>% combine_words()
)
)
}
descri.text
[1] "CD4: T cells CD4 naive, T cells CD4 memory resting, T cells CD4 memory activated, T cells follicular helper, and T cells regulatory (Tregs)"
[2] "CD8: T cells CD8"
[3] "CD14: Monocytes, Macrophages M0, Macrophages M1, and Macrophages M2"
[4] "CD19: B cells naive and B cells memory"
[5] "PCs: Plasma cells"
[6] "GammaT: T cells gamma delta"
[7] "NK: NK cells resting and NK cells activated"
[8] "Dendritic: Dendritic cells resting and Dendritic cells activated"
[9] "Mast: Mast cells resting and Mast cells activated"
[10] "Eos: Eosinophils"
[11] "PMN: Neutrophils"
gpar(fontface = "bold")
$font
bold
2
## https://github.com/wilkelab/gridtext/issues/24
update_gpar <- function(gp, gp_new) {
names_new <- names(gp_new)
names_old <- names(gp)
gp[c(intersect(names_old, names_new), "fontface")] <- NULL
gp_new["fontface"] <- NULL
do.call(gpar, c(gp, gp_new))
}
gp <- gpar(fontface = "bold")
gpmod <- update_gpar(gp, gpar(fontsize = 10))
## heatmaps for inbuilt and custom
ht_sigmtx <- lapply(names(sigmtxs), function(nin) {
datmtx <- sigmtxs[[nin]]$pltmat %>%
pheatmap:::scale_mat(., "row")
datcol <- sigmtxs[[nin]]$col_fun
datsplit <- sigmtxs[[nin]]$matorder
Heatmap(
matrix = datmtx, col = datcol,
cluster_columns = FALSE,
show_row_names = FALSE,
column_split = datsplit,
column_gap = unit(1, "mm"),
## column_title_gp = gpar(fontface = "bold",
## fontsize=10),
column_title_gp = gpmod,
name = paste0(nin, "\nExpr")
)
})
names(ht_sigmtx) <- names(sigmtxs)Code
## observed expr
observ_expr <- file.path(
data_dir,
"CLUSTERData/ObservTPMExpr.rds"
) %>%
readRDS()
########################################################
## sig genes in observ_expr, including debCAM markers ##
########################################################
## cell type train and testing samples
sampin4pca <- sampdata[celltype != "PBMC" & samtype == "nonrefsamp", sample]
## sampin4pca <- sampdata[celltype!="PBMC", sample]
pca1_4res <- list(
inbuilt = rownames(sigmtxs$inbuilt$pltmat),
custom = rownames(sigmtxs$custom$pltmat),
debCAM = unlist(debCAMresFract$OVE.FC_10$markers, use.names = FALSE)
) %>% lapply(., function(sgin) {
## some LM22 genes are not found in observ_expr
gin <- intersect(sgin, rownames(observ_expr))
## pca of signature genes in our observed expr
mat <- log2(observ_expr[gin, sampin4pca] + 1)
mat %>% dim()
pca_res <- prcomp(t(mat))
pcs <- pca_res$x[, seq(1, 4)] %>% as.data.frame()
pcs$celltype <- sampdata$celltype[match(rownames(pcs), sampdata$sample)]
pcs$celltype <- factor(pcs$celltype, levels = sctorder)
pcs$Batch <- sampdata$batch[match(rownames(pcs), sampdata$sample)]
pcs$Batch <- factor(pcs$Batch)
pcaplts <- lapply(1:2, function(x) {
x1 <- paste0("PC", 2 * x - 1)
y1 <- paste0("PC", 2 * x)
ggscatter(pcs,
x = x1, y = y1,
fill = "celltype",
shape = "Batch",
alpha = 0.9,
ellipse = TRUE,
ellipse.type = "convex",
ellipse.alpha = 0.3,
ellipse.border.remove = TRUE,
palette = ctfrat.pal
) +
scale_shape_manual(
values = c(
"1" = 21,
"2" = 22,
"3" = 23,
"4" = 24
),
limits = force
) +
guides(fill = guide_legend(override.aes = list(shape = 21))) +
used_ggthemes()
}) %>% ggarrange(plotlist = ., common.legend = TRUE)
list(
gin = gin,
pcaplts = pcaplts
)
})
figS10def <- list(
inbuilt = rownames(sigmtxs$inbuilt$pltmat),
custom = rownames(sigmtxs$custom$pltmat),
debCAM = unlist(debCAMresFract$OVE.FC_10$markers, use.names = FALSE)
) %>% lapply(., function(sgin) {
## some LM22 genes are not found in observ_expr
gin <- intersect(sgin, rownames(observ_expr))
## pca of signature genes in our observed expr
mat <- log2(observ_expr[gin, sampin4pca] + 1)
mat %>% dim()
pca_res <- prcomp(t(mat))
pcs <- pca_res$x[, seq(1, 4)] %>% as.data.frame()
pcs$celltype <- sampdata$celltype[match(rownames(pcs), sampdata$sample)]
pcs$celltype <- factor(pcs$celltype, levels = sctorder)
pcs$Batch <- sampdata$batch[match(rownames(pcs), sampdata$sample)]
pcs$Batch <- factor(pcs$Batch)
pcs
})
names(figS10def) <- paste0("figS10", c("d", "e", "f"))- Expression of inbuilt signature genes (N=547) in 22 leukocyte subsets, curated by the CIBERSORTx team from microarray gene expression. For each gene (row), expression is centred to the mean and scaled by the standard deviation of cell types (column). Columns are split by the collapsed classes. CD4: T cells CD4 naive, T cells CD4 memory resting, T cells CD4 memory activated, T cells follicular helper, and T cells regulatory (Tregs); CD8: T cells CD8; CD14: Monocytes, Macrophages M0, Macrophages M1, and Macrophages M2; CD19: B cells naive and B cells memory; PCs: Plasma cells; GammaT: T cells gamma delta; NK: NK cells resting and NK cells activated; Dendritic: Dendritic cells resting and Dendritic cells activated; Mast: Mast cells resting and Mast cells activated; Eos: EosinophilsPMN: Neutrophils
Code
## inbuilt signature genes
ht_sigmtx[[1]]
- Expression of custom signature genes (N=1589), derived from our sorted-cell RNAseq expression in 80 training subjects using CIBERSORTx. Expression is centred and scaled across cell types (column) by gene (row).
Code
## custom signature genes
ht_sigmtx[[2]]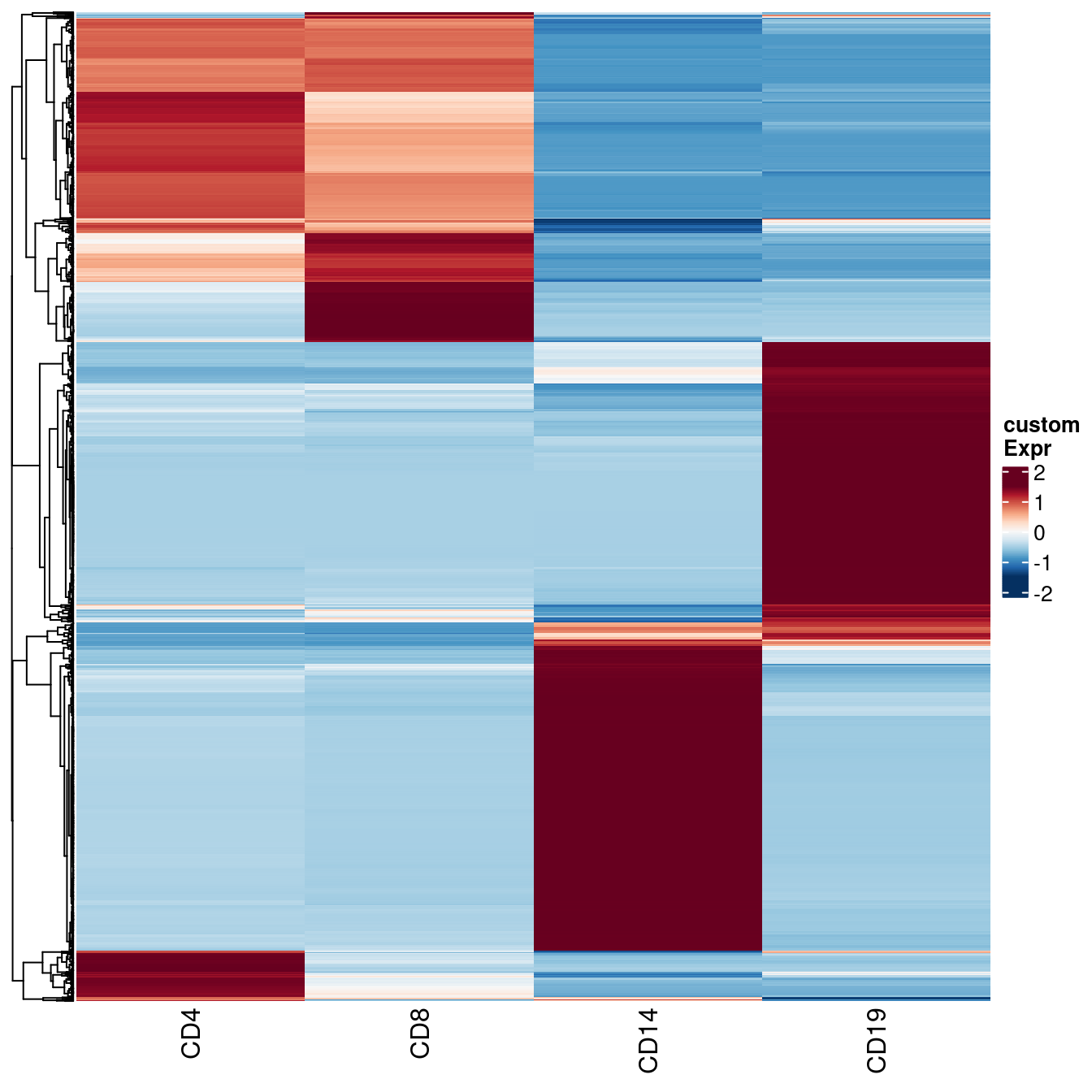
- Numbers (No.) of debCAM cell-type specific genes by cell type (row) and selection criteria (column). debCAM, which does not have the signature matrix, selects the cell-type-specific genes, that are over-expressed (OVE) in one cell type compared to others combined. OVE fold change (FC) of 1, 2, 5 and 10 were used in debCAM on our sorted cell expression in 80 training subjects.
Code
## debCAM cell type-specific genes by fold changes
## supple for debCAM
## summary of no. markers by 4 FCs
debCAMmarkers <- lapply(debCAMresFract, function(dat) {
sapply(dat$markers, function(x) {
length(x)
})
}) %>%
do.call(rbind, .) %>%
as.data.frame() %>%
setDT(., keep.rownames = TRUE)
## no. markers by fc from debcam
debbarplt <- melt(debCAMmarkers, id.vars = "rn", measure.vars = sctorder, variable.name = "celltype", variable.factor = sctorder)[, rn := factor(rn, names(debCAMresFract))] %>%
ggplot(data = ., aes(x = celltype, y = value)) +
geom_bar(stat = "identity", fill = "#69b3a2", width = 0.8) +
geom_text(aes(label = value, hjust = ifelse(value < 500, 0, 0.8)), size = 3.5) +
coord_flip() +
facet_wrap(~rn, ncol = 4) +
ylab("No. cell-type specific genes") +
xlab("cell type") +
used_ggthemes()
debbarplt
figS10c <- melt(debCAMmarkers, id.vars = "rn", measure.vars = sctorder, variable.name = "celltype", variable.factor = sctorder)[, rn := factor(rn, names(debCAMresFract))]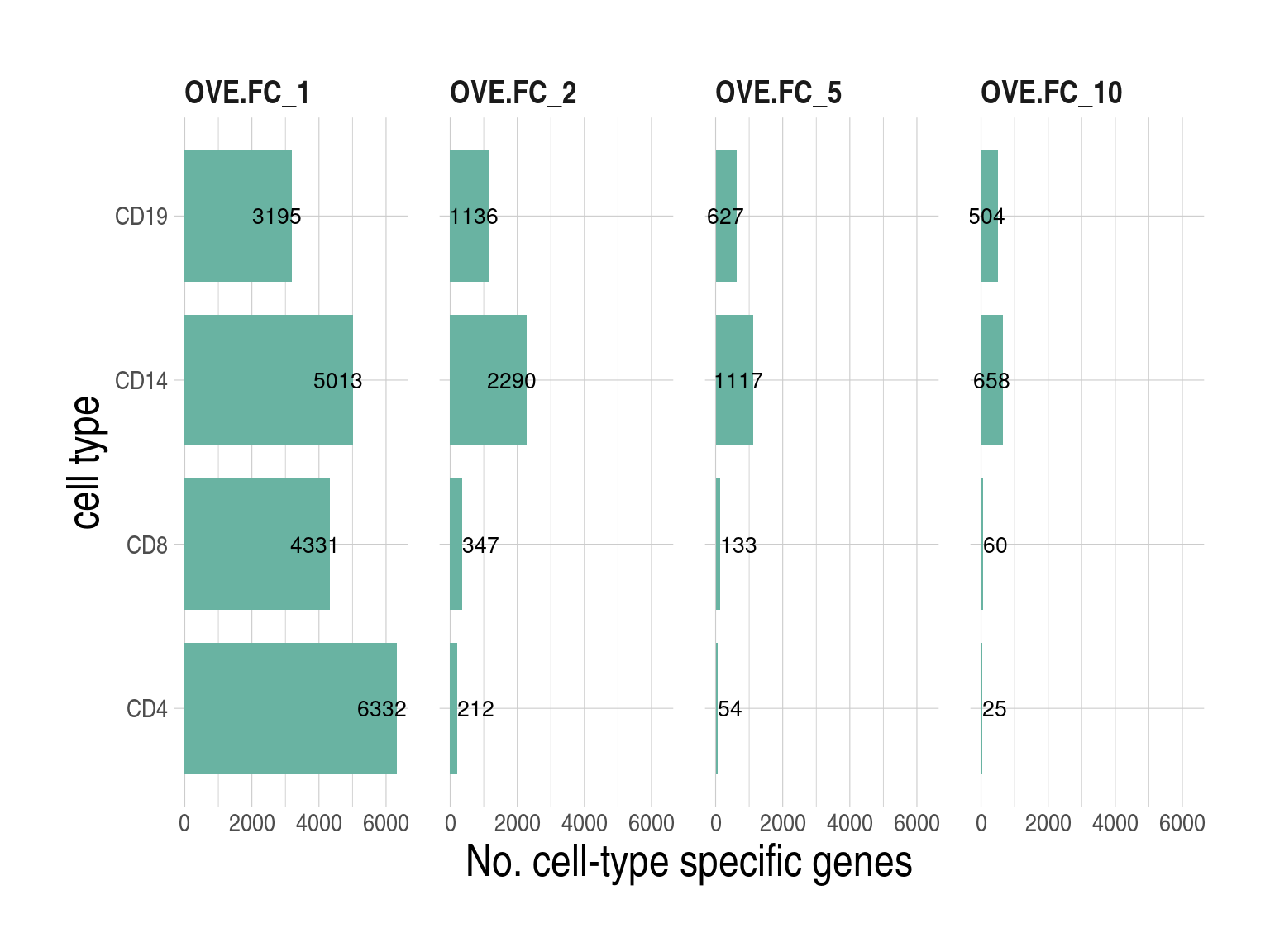
- The first four principal components from PCA analysis of inbuilt signature gene expression in test samples. Each dot is a sample, coloured by cell type and shaped by sequencing batch.
Code
## PCA analysis of cell type expression data in test samples
### inbuilt signature genes
pca1_4res$inbuilt$pcaplts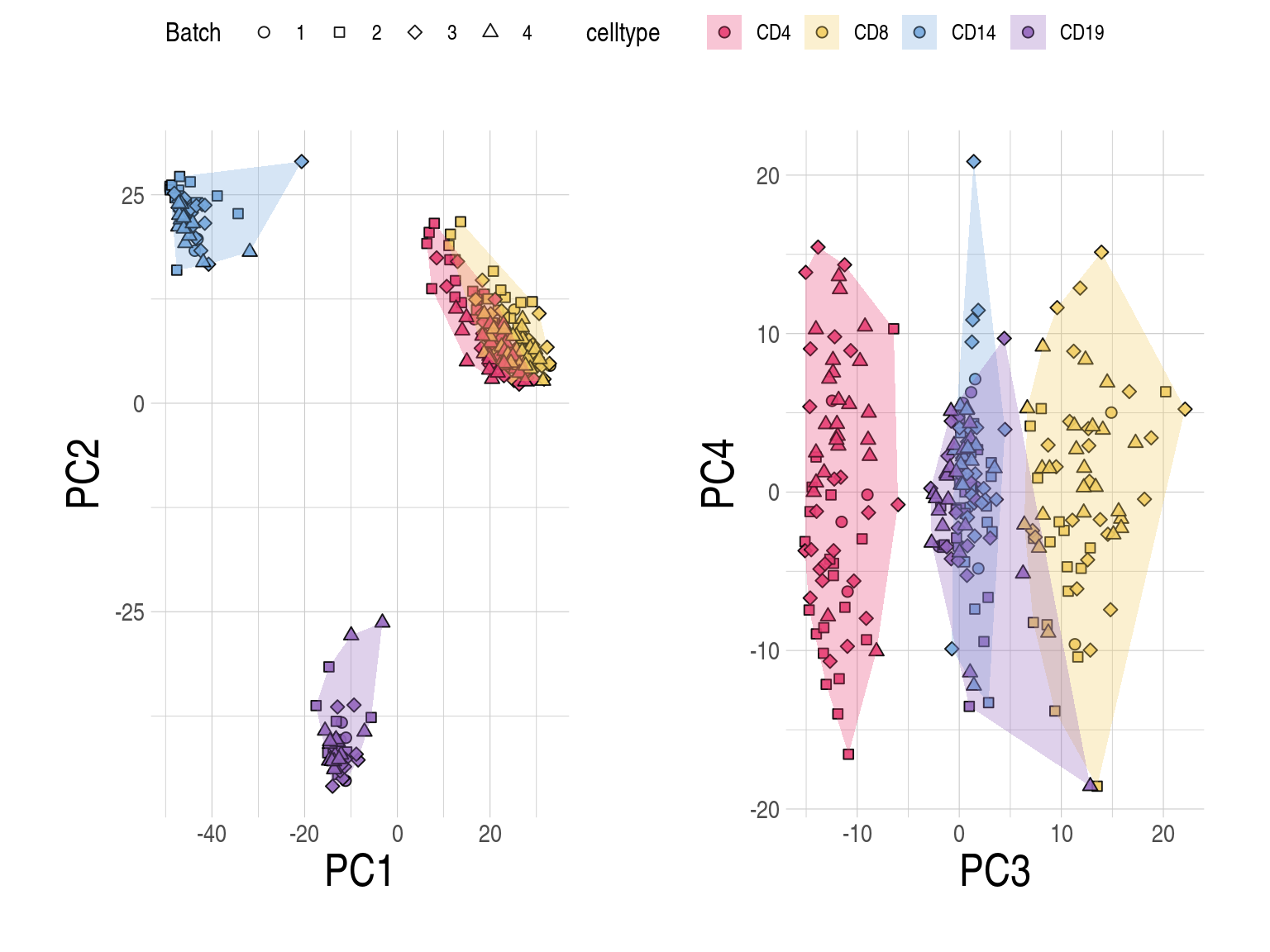
- The first four principal components from PCA analysis of custom signature gene expression in test samples.
Code
### custum signature genes
pca1_4res$custom$pcaplts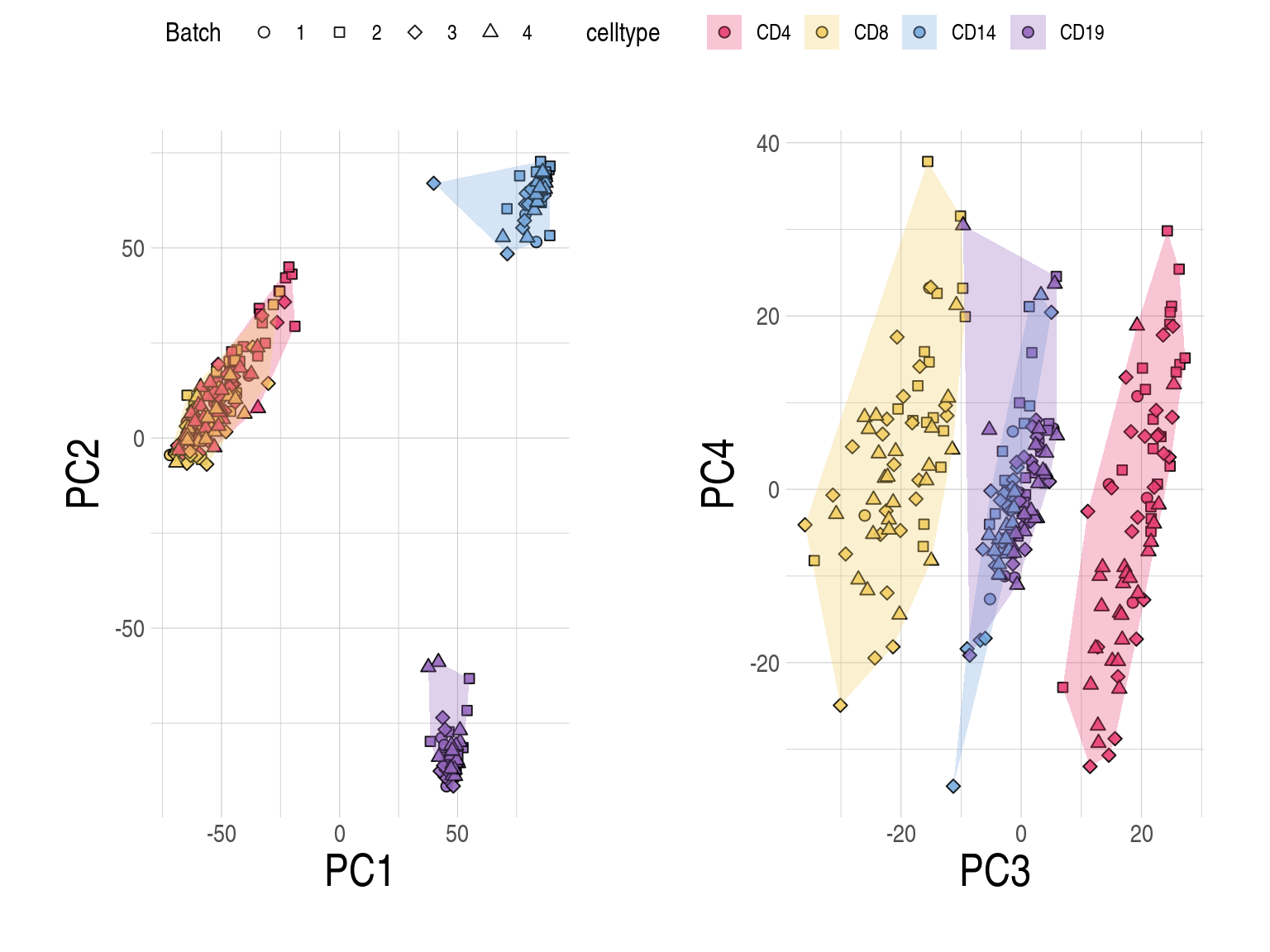
- The first four principal components from PCA analysis of debCAM cell-type specific gene expression in test samples.
Code
### debCAM cell-type specific genes
pca1_4res$debCAM$pcaplts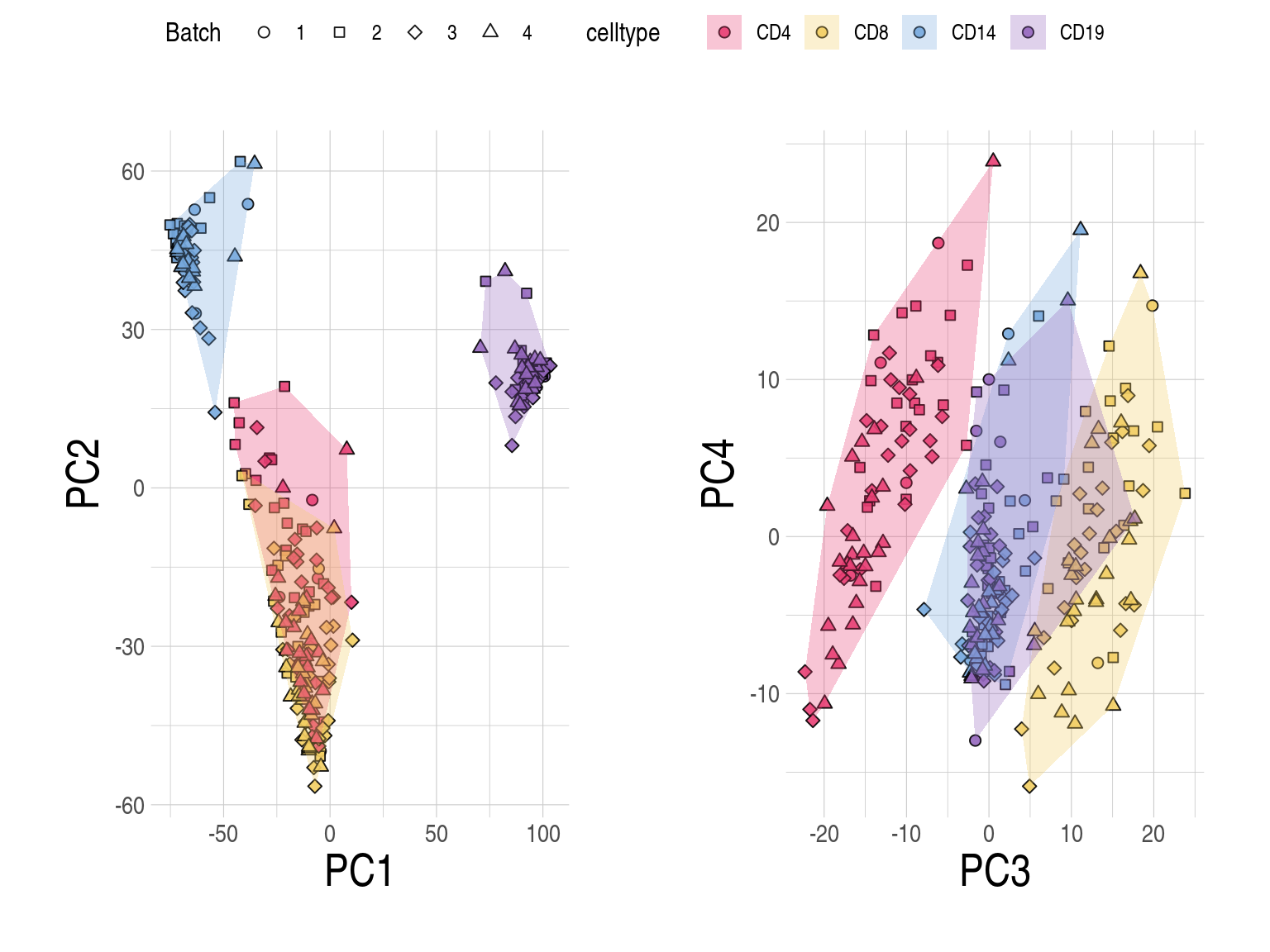
2.11 SupFig12, Combat-seq
- The first four principal components from PCA analysis of log2(count-per-million) expression derived from raw read counts in RNAseq samples used in this work.
Code
res_d <- file.path(data_dir, "ZenodoCLUSTER")
rawreadtbl <- read.table(file.path(res_d, "RawReadCounts_Zenodo.csv"), row.names = 1, sep = ",", header = TRUE)
rawreadtbl <- rawreadtbl[, sampdata$sample]
adjreadtbl <- file.path(res_d, "BatchCorrectedReadCounts_Zenodo.csv") %>%
read.table(file = ., row.names = 1, sep = ",", header = TRUE)
adjreadtbl <- adjreadtbl[, sampdata$sample]
geneMeta <- file.path(res_d, "GeneMetaInfo_Zenodo.csv") %>%
read.table(
file = ., row.names = 1, sep = ",",
header = TRUE
)
pca_res <- list(rawreadtbl, adjreadtbl) %>%
lapply(., function(countmtx) {
objinput <- DGEList(
counts = countmtx,
genes = geneMeta[match(rownames(countmtx), geneMeta$Geneid), ]
)
keep <- filterByExpr(objinput,
group = sampdata$celltype[match(colnames(objinput), sampdata$sample)]
)
objinput <- objinput[keep, , keep.lib.sizes = FALSE]
objinput <- calcNormFactors(objinput)
matcpm <- cpm(objinput, log = TRUE)
respca <- prcomp(x = t(matcpm))
stopifnot(all(rownames(respca$x) == sampdata$sample))
dsn4pca <- cbind(respca$x[, 1:4], sampdata[, .(celltype, batch)])
dsn4pca$batch <- factor(dsn4pca$batch, levels = 1:4)
dsn4pca$celltype <- factor(
dsn4pca$celltype,
levels = c("PBMC", sctorder)
)
dsn4pca
})
figS12ab <- copy(pca_res)
names(figS12ab) <- paste0("figS12", letters[seq_along(pca_res)])
ctfrat.palpmbc <- c(ctfrat.pal, "#AAAA00")
names(ctfrat.palpmbc) <- c(sctorder, "PBMC")
pca_plts <- lapply(pca_res, function(datin) {
pcaplts <- lapply(1:2, function(x) {
x1 <- paste0("PC", 2 * x - 1)
y1 <- paste0("PC", 2 * x)
ggscatter(datin,
x = x1, y = y1,
fill = "celltype",
shape = "batch",
alpha = 0.9,
palette = ctfrat.palpmbc
) +
scale_shape_manual(
values = c(
"1" = 21,
"2" = 22,
"3" = 23,
"4" = 24
),
limits = force
) +
guides(fill = guide_legend(override.aes = list(shape = 21))) +
used_ggthemes()
})
})
## raw
ggarrange(plotlist = pca_plts[[1]], common.legend = TRUE)
- The first four principal components from PCA analysis of log2(count-per-million) expression derived from Combat-Seq batch-adjusted read counts in RNAseq samples used in this work
Code
## combat-seq
ggarrange(plotlist = pca_plts[[2]], common.legend = TRUE)
2.12 SupFig13, Batch composition of subjects by train and test set
- Frequencies (A) and percentages (B) of subjects by batch and training/testing set.
Code
## Batch composition of subjects by train and test set
categoryplt <- function(dat, xgroup, fillvar, ...) {
## count
adsn <- dat[!is.na(get(xgroup)), .N, by = c(xgroup, fillvar)][eval(parse(text = paste0("order(", xgroup, ",", fillvar, ")")))]
ares <- adsn %>% ggbarplot(
data = ., x = xgroup, y = "N",
fill = fillvar,
ylab = "Counts", ...
)
## percentage
bdsn <- dat[!is.na(get(xgroup)), .N, by = c(xgroup, fillvar)][eval(parse(text = paste0("order(", xgroup, ",", fillvar, ")")))]
bdsn <- bdsn[,
{
an <- sum(N)
props <- N / an
labelt <- round(props, 2)
list(eval(parse(text = fillvar)), N, an, props, labelt)
},
by = xgroup
]
bres <- bdsn %>% ggbarplot(
data = ., x = xgroup, y = "labelt", fill = "V1",
ylab = "Percentages", ...
)
bres
overp <- chisq.test(dat[[fillvar]], dat[[xgroup]])$p.value
overp <- ifelse(overp < 10^-2,
paste0("=", format(overp,
nsmall = 2,
digits = 3,
scientific = TRUE
)),
paste0("=", round(overp, 2))
)
if (class(dat[[xgroup]]) == "character") {
dat[[xgroup]] <- factor(dat[[xgroup]])
}
## grp <- unique(dat[[xgroup]])
grp <- levels(dat[[xgroup]])
dddf <- data.table(
group1 = grp[1], group2 = grp[length(grp)],
p = paste0(" p ", overp)
)
bres <- bres +
stat_pvalue_manual(dddf,
y.position = 1.02,
bracket.nudge.y = 0.02,
label = "Chi-square test, {p}"
) +
labs(fill = fillvar)
list(ares, bres)
}
## subjects by train and test set
sampdata.sub <- unique(sampdata, by = "samp")
sampdata.sub[, Batch := factor(batch, levels = seq(1, 4))]
sampdata.sub[, sample_type := factor(samtype, levels = c("refsamp", "nonrefsamp"), labels = c("train", "Test"))]
categoryplt(
dat = sampdata.sub,
xgroup = "sample_type",
fillvar = "Batch",
xlab = FALSE,
label = TRUE,
lab.pos = "in",
palette = "jama"
) %>%
ggarrange(plotlist = ., common.legend = TRUE, labels = LETTERS)
figS13 <- sampdata.sub[, .(samp, sample_type, Batch)]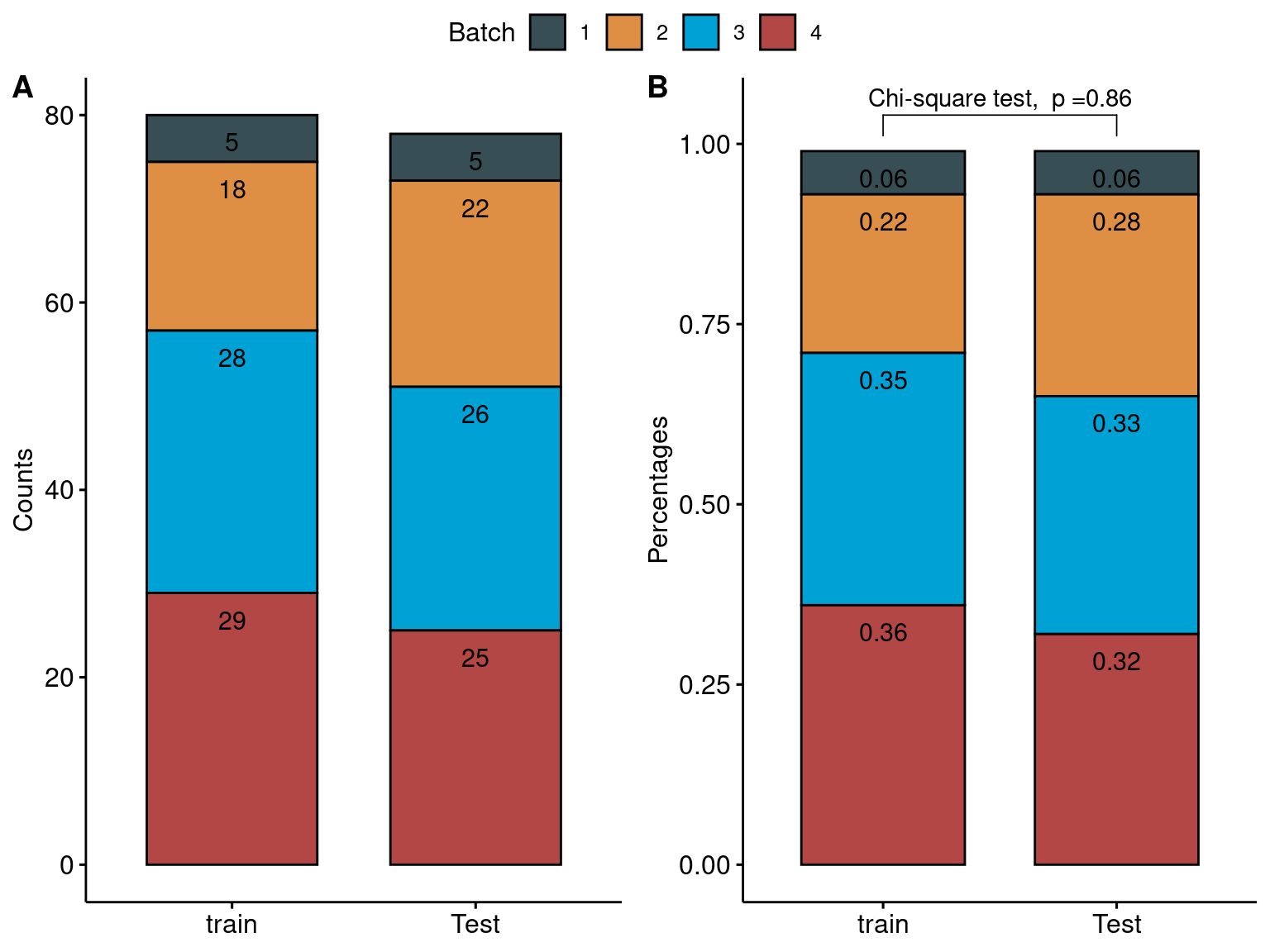
2.13 SupFig14, Correalations of fractions between inbuilt cell linages with true flow data
Code
## Fractions between 22 inbuilt cell lineages with flow data
####################
## flow fractions ##
####################
fractflow <- file.path(data_dir, "CLUSTERData/flowFract_wide.rds") %>%
readRDS() %>%
as.matrix()
## cibx-LM22
#############################
## fractions based on LM22 ##
#############################
lm22merged <- "data4repo/mergedClasses.csv" %>%
file.path(here::here(), .) %>%
scan(., what = "character", sep = "\t")
lm22mtx <- lm22mtx_f %>% fread()
res_d <- file.path("CIBXres/LM22deconv", runid) %>%
file.path(data_dir, .)
res_d
[1] "/rds/project/rds-csoP2nj6Y6Y/wyl37/Rbookdown/sceQTLGen/CLUSTERdeconRes/CIBXres/LM22deconv/cluster"
lm22res <- file.path(res_d, "CIBERSORTxGEP_NA_Fractions-Adjusted.txt") %>%
fread()
stopifnot(names(lm22res)[-1][1:length(names(lm22mtx)[-1])] == names(lm22mtx)[-1])
## fractions in 22 cell types
lm22res.mtx <- lm22res[, names(lm22mtx)[-1], with = FALSE] %>% as.matrix()
rownames(lm22res.mtx) <- lm22res$Mixture
ourorder <- lm22merged
names(ourorder) <- names(lm22mtx)[-1]
othercls <- setdiff(unique(ourorder), sctorder)
ourorder <- sort(factor(ourorder, levels = c(sctorder, othercls)))
## sample order matched to fractflow
lm22res.mtx <- lm22res.mtx[rownames(fractflow), names(ourorder)]
## test samples
nonrefsamps <- sampdata[samtype == "nonrefsamp" & celltype == "PBMC", sample]
## correlation
mychks_test <- cor(
x = fractflow[nonrefsamps, ],
y = lm22res.mtx[nonrefsamps, ]
)
annotcoldf <- data.frame(mergedClass = ourorder)
rownames(annotcoldf) <- names(ourorder)
## find column split
kkkres <- S4Vectors::Rle(ourorder)
annotcol_pal <- friend11
names(annotcol_pal) <- levels(ourorder)
annotcol_pal_dat <- list(mergedClass = annotcol_pal)
## heatmap correlation
library(circlize)
col_fun <- circlize::colorRamp2(
c(1, 0, -1),
colorspace::diverging_hcl(palette = "Red-Green", n = 3)
)
gp <- gpar(fontface = "bold")
update_gpar(gp, gpar(fontsize = 10))
$font
[1] 2
$fontsize
[1] 10
beforeCorht <- ComplexHeatmap::pheatmap(
mat = mychks_test,
name = "Cor",
color = col_fun,
cluster_rows = FALSE, cluster_cols = FALSE,
display_numbers = TRUE, number_format = "%.2f",
number_color = "black",
annotation_col = annotcoldf,
annotation_colors = annotcol_pal_dat,
gaps_col = S4Vectors::end(kkkres),
cellwidth = 25, cellheight = 25,
row_names_side = "left"
)
figS14 <- mychks_test- Pearson correlations between inbuilt LM22 and ground-truth flow fractions. Columns are LM22 cell subsets split by their merged classes (top annotation). Rows are ground truth cell types. Each cell is coloured based on the strength of the correlation.
Code
beforeCorht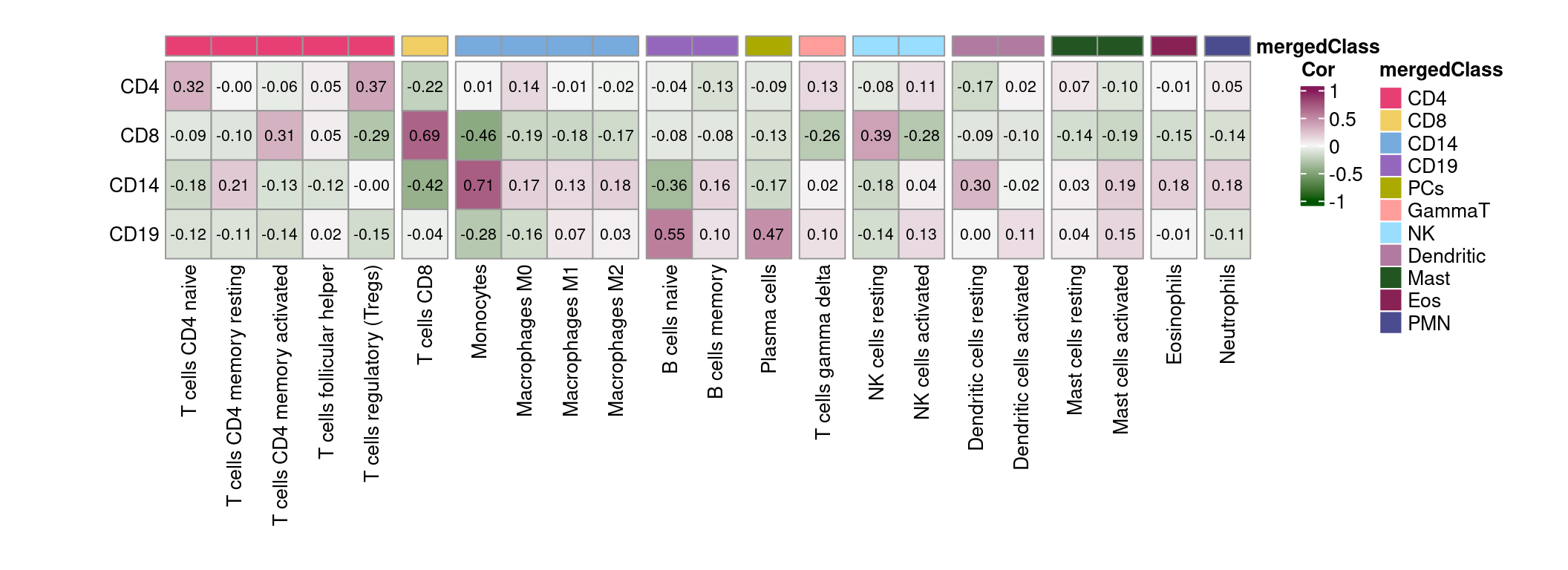
Code
ddd <- ls(pattern = "figS") %>% str_sort(., numeric = TRUE)
dddidx <- sapply(ddd, get) %>% sapply(., function(x) inherits(x, "list"))
cccin <- ddd[!dddidx]
ccc <- vector(mode = "list", length = length(cccin))
for (i in seq_along(cccin)) {
ccc[[i]] <- get(cccin[i])
}
names(ccc) <- cccin
ddd[dddidx]
[1] "figS1" "figS10ab" "figS10def" "figS12ab"
outlist <- c(
figS1, ccc[1:12],
figS10ab, ccc["figS10c"],
figS12ab, ccc[14:15]
)
names(outlist) <- toupper(names(outlist)) %>%
str_extract(., pattern = "S.+$") %>%
paste0(., "_fig")
openxlsx::write.xlsx(outlist,
file = "SourceData_SuppFigures.xlsx"
)2.14 AUCs in Test samples, CLUSTER data
Code
rds_f <- "fig4rundatatestonly.rds"
if (!file.exists(rds_f)) {
## main: different gene set; common: same genes per cell across methods
## impvexpr_all <- list(main = expr_ori, common = expr_scenarios)
impvexpr_all <- list(
main = cdata_explst$iexpr,
common = cdata_explst$icexpr
)
## TRUE for dichotomous; FALSE for continuous
dcht_pheno <- c(TRUE, FALSE)
names(dcht_pheno) <- c("dcht", "cntn")
numCores <- length(expr_order)
numCores
cl <- makeCluster(numCores, type = "FORK")
registerDoParallel(cl)
aucall <- foreach(
j = seq_along(impvexpr_all),
.final = function(x) setNames(x, names(impvexpr_all))
) %:%
foreach(
i = dcht_pheno,
.final = function(x) setNames(x, names(dcht_pheno))
) %dopar% {
observy <- cdata_explst$oexpr
train_samps_ct <- cdata_explst$trainsamp
sampdata <- cdata_explst$phenodata
dat <- run_datprepfig4(
obvexpr = observy,
impvexpr = impvexpr_all[[j]],
tsampct = NULL,
dich = i
)
## expr ~ pheno
res1 <- run_fig4(datprlst = dat, truesig = 0.05)
## expr~ sex + pheno
res2 <- run_fig4(
datprlst = dat, truesig = 0.05,
covdata = sampdata, covid = "sample", covar = "sex"
)
list(nocov = res1, covin = res2)
}
stopCluster(cl)
mychktestonly <- unlist(aucall, recursive = FALSE) %>%
unlist(., recursive = FALSE) %>%
lapply(., "[[", "aucres") %>%
lapply(., function(datin) {
datin[, ":="(
celltype = factor(celltype, levels = sctorder),
approach = factor(approach, levels = expr_order))]
datin
}) %>%
rbindlist(l = ., idcol = "scenarios") %>%
.[, pheno := gsub("^main\\.|^common\\.", "", scenarios)] %>%
.[, scenario := str_extract(scenarios, "main|common")]
saveRDS(mychktestonly, file = rds_f)
} else {
mychktestonly <- readRDS(rds_f)
}2.14.1 Varying gene sets
Code
pheno.order <- CJ(c("dcht", "cntn"), c("nocov", "covin"), sorted = FALSE)[, paste(V1, V2, sep = ".")]
pheno.order
[1] "dcht.nocov" "dcht.covin" "cntn.nocov" "cntn.covin"
mychktestonly[, pheno := factor(pheno, levels = pheno.order, labels = c("dichPheno", "dichPheno+sex", "contPheno", "contPheno+sex"))]
## no. auc available
achk <- mychktestonly[scenario == "main" & !is.na(auc), .N, by = c("pheno", "celltype", "approach")][N != 10, ]
achk
pheno celltype approach N
1: dichPheno CD8 inbuilt 8
2: dichPheno CD8 custom 8
3: dichPheno CD8 bMIND 9
4: dichPheno CD8 swCAM 9
5: dichPheno CD8 LASSO 9
6: dichPheno CD8 RIDGE 9
7: dichPheno CD14 inbuilt 7
8: dichPheno CD14 custom 8
9: dichPheno CD14 bMIND 7
10: dichPheno CD14 swCAM 7
11: dichPheno CD14 LASSO 7
12: dichPheno CD14 RIDGE 7
13: dichPheno CD19 inbuilt 8
14: dichPheno CD19 custom 8
15: dichPheno CD19 bMIND 8
16: dichPheno CD19 swCAM 8
17: dichPheno CD19 LASSO 8
18: dichPheno CD19 RIDGE 8
19: dichPheno+sex CD4 inbuilt 9
20: dichPheno+sex CD4 custom 8
21: dichPheno+sex CD8 inbuilt 7
22: dichPheno+sex CD8 custom 7
23: dichPheno+sex CD8 bMIND 8
24: dichPheno+sex CD8 swCAM 8
25: dichPheno+sex CD8 LASSO 8
26: dichPheno+sex CD8 RIDGE 8
27: dichPheno+sex CD14 inbuilt 8
28: dichPheno+sex CD14 custom 8
29: dichPheno+sex CD14 bMIND 7
30: dichPheno+sex CD14 swCAM 7
31: dichPheno+sex CD14 LASSO 7
32: dichPheno+sex CD14 RIDGE 7
33: dichPheno+sex CD19 inbuilt 8
34: dichPheno+sex CD19 custom 8
35: dichPheno+sex CD19 bMIND 9
36: dichPheno+sex CD19 swCAM 9
37: dichPheno+sex CD19 LASSO 9
38: dichPheno+sex CD19 RIDGE 9
39: contPheno CD8 inbuilt 9
40: contPheno CD14 inbuilt 9
41: contPheno CD14 custom 9
42: contPheno CD14 bMIND 9
43: contPheno CD14 swCAM 9
44: contPheno CD14 LASSO 8
45: contPheno CD14 RIDGE 9
46: contPheno+sex CD14 inbuilt 9
47: contPheno+sex CD14 custom 9
48: contPheno+sex CD14 LASSO 9
49: contPheno+sex CD19 inbuilt 9
pheno celltype approach N
achk_words <- "**Noted AUC was not calculated for some simulated phenotypes because all associations were true negative, and none were false negative. Scenario-celltype-approach with no. AUCs $<$ 10 are listed in the code chunk.**"
ggboxplot(
data = mychktestonly[scenario == "main", ],
x = "approach", y = "auc", ylab = "AUC",
palette = appro.cols,
alpha = 0.35, fill = "approach",
add = c("jitter"),
add.params = list(shape = 21, size = 2.5, alpha = 1)
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(pheno ~ celltype, scales = "free") +
labs(fill = NULL) +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "top",
strip.text.y.right = element_text(angle = 0),
legend.text = element_text(size = rel(1))
) +
rremove("xlab")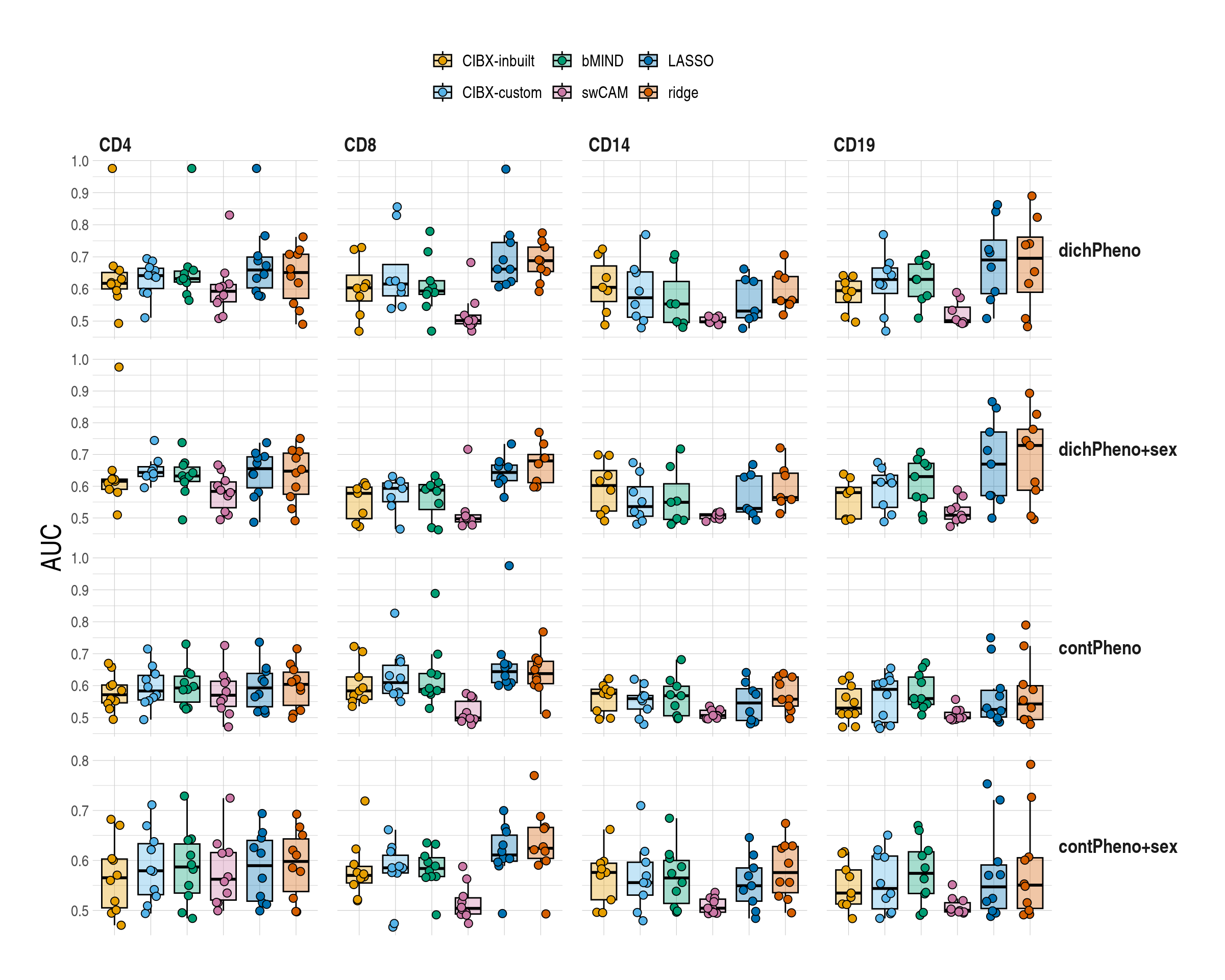
Code
mychktestonly_tbl <- copy(mychktestonly)
mychktestonly_tbl[, .N, by = c("scenarios", "celltype", "approach")]
scenarios celltype approach N
1: main.dcht.nocov CD4 inbuilt 10
2: main.dcht.nocov CD4 custom 10
3: main.dcht.nocov CD4 bMIND 10
4: main.dcht.nocov CD4 swCAM 10
5: main.dcht.nocov CD4 LASSO 10
---
188: common.cntn.covin CD19 custom 10
189: common.cntn.covin CD19 bMIND 10
190: common.cntn.covin CD19 swCAM 10
191: common.cntn.covin CD19 LASSO 10
192: common.cntn.covin CD19 RIDGE 10
tbl_summary <- mychktestonly_tbl[, list(
Q50 = median(auc, na.rm = TRUE) %>% round(., 2),
Q25 = quantile(auc, probs = 0.25, na.rm = TRUE) %>% round(., 2),
Q75 = quantile(auc, probs = 0.75, na.rm = TRUE) %>% round(., 2)
),
by = c("scenarios", "celltype", "approach")
][
, V1 := paste0(Q50, " (", Q25, "-", Q75, ")")
]
tbl_summary[, pheno := gsub("^main\\.|^common\\.", "", scenarios)]
tbl_summary[, scenario := str_extract(scenarios, "main|common")]
tbl_res <- dcast(
data = tbl_summary, pheno + celltype ~ scenario + approach,
value.var = "V1"
)
setDF(tbl_res, rownames = paste0(tbl_res$pheno, tbl_res$celltype))
pheno.order <- CJ(c("dcht", "cntn"), c("nocov", "covin"), sorted = FALSE)[, paste(V1, V2, sep = ".")]
cols.order <- CJ(c("main", "common"), V2 = expr_order, sorted = FALSE)[, paste(V1, V2, sep = "_")]
rows.order <- CJ(pheno.order, sctorder, sorted = FALSE)[, paste0(pheno.order, sctorder)]
tbl_out <- tbl_res[rows.order, c("celltype", grep("main", cols.order, value = TRUE))]
names(tbl_out) <- gsub("main_|common_", "", names(tbl_out))
capin <- paste0(
"Median AUC (Q25-Q75) across",
ifelse(nrow(achk) != 0, "", uniqueN(mychktestonly$fakedPCs)),
" simulated phentoypes per cell by apparoch. **Genes common across approaches per cell type**"
)
kbl(
x = tbl_out, row.names = FALSE,
caption = capin
) %>%
kable_paper("striped", full_width = FALSE) %>%
pack_rows("dichotomous pheno", 1, 4,
label_row_css = "text-align: left;"
) %>%
pack_rows("dichotomous pheno + sex", 5, 8) %>%
pack_rows("continous pheno", 9, 12) %>%
pack_rows("continous pheno + sex", 13, 16) %>%
column_spec(column = 6:7, bold = T)| celltype | inbuilt | custom | bMIND | swCAM | LASSO | RIDGE |
|---|---|---|---|---|---|---|
| dichotomous pheno | ||||||
| CD4 | 0.62 (0.6-0.65) | 0.64 (0.6-0.66) | 0.63 (0.62-0.66) | 0.59 (0.56-0.61) | 0.66 (0.6-0.7) | 0.65 (0.57-0.71) |
| CD8 | 0.6 (0.56-0.64) | 0.62 (0.58-0.68) | 0.59 (0.58-0.63) | 0.5 (0.49-0.52) | 0.66 (0.62-0.74) | 0.69 (0.65-0.73) |
| CD14 | 0.61 (0.56-0.67) | 0.57 (0.51-0.65) | 0.55 (0.5-0.62) | 0.5 (0.5-0.51) | 0.53 (0.51-0.63) | 0.56 (0.56-0.64) |
| CD19 | 0.59 (0.56-0.62) | 0.63 (0.59-0.67) | 0.63 (0.58-0.68) | 0.5 (0.5-0.54) | 0.69 (0.59-0.75) | 0.7 (0.59-0.76) |
| dichotomous pheno + sex | ||||||
| CD4 | 0.62 (0.59-0.62) | 0.64 (0.63-0.66) | 0.63 (0.61-0.66) | 0.58 (0.53-0.61) | 0.66 (0.59-0.69) | 0.65 (0.57-0.7) |
| CD8 | 0.58 (0.5-0.6) | 0.59 (0.55-0.61) | 0.59 (0.53-0.61) | 0.5 (0.49-0.51) | 0.64 (0.62-0.67) | 0.68 (0.61-0.7) |
| CD14 | 0.6 (0.52-0.65) | 0.54 (0.51-0.6) | 0.55 (0.5-0.61) | 0.51 (0.5-0.51) | 0.53 (0.52-0.63) | 0.56 (0.56-0.64) |
| CD19 | 0.58 (0.5-0.6) | 0.61 (0.53-0.63) | 0.63 (0.56-0.67) | 0.51 (0.5-0.53) | 0.67 (0.57-0.77) | 0.73 (0.59-0.78) |
| continous pheno | ||||||
| CD4 | 0.57 (0.55-0.6) | 0.58 (0.56-0.63) | 0.59 (0.55-0.63) | 0.57 (0.54-0.61) | 0.59 (0.53-0.64) | 0.6 (0.54-0.64) |
| CD8 | 0.58 (0.56-0.63) | 0.61 (0.58-0.66) | 0.59 (0.58-0.64) | 0.5 (0.49-0.55) | 0.64 (0.61-0.67) | 0.64 (0.61-0.68) |
| CD14 | 0.58 (0.52-0.59) | 0.56 (0.53-0.57) | 0.57 (0.51-0.6) | 0.51 (0.5-0.52) | 0.55 (0.49-0.59) | 0.56 (0.54-0.63) |
| CD19 | 0.53 (0.51-0.59) | 0.59 (0.48-0.61) | 0.56 (0.54-0.63) | 0.5 (0.5-0.52) | 0.53 (0.5-0.59) | 0.54 (0.49-0.6) |
| continous pheno + sex | ||||||
| CD4 | 0.57 (0.51-0.6) | 0.58 (0.53-0.63) | 0.59 (0.53-0.63) | 0.56 (0.52-0.62) | 0.59 (0.52-0.64) | 0.6 (0.54-0.64) |
| CD8 | 0.57 (0.55-0.59) | 0.59 (0.57-0.61) | 0.58 (0.57-0.61) | 0.5 (0.49-0.53) | 0.61 (0.6-0.65) | 0.62 (0.6-0.67) |
| CD14 | 0.58 (0.52-0.59) | 0.56 (0.53-0.6) | 0.56 (0.51-0.6) | 0.5 (0.5-0.52) | 0.55 (0.52-0.58) | 0.58 (0.54-0.63) |
| CD19 | 0.53 (0.51-0.58) | 0.54 (0.5-0.61) | 0.57 (0.53-0.62) | 0.5 (0.5-0.52) | 0.55 (0.5-0.59) | 0.55 (0.5-0.61) |
2.14.2 Common gene sets
Code
achk1 <- mychktestonly[scenario == "common" & !is.na(auc), .N, by = c("pheno", "celltype", "approach")][N != 10, ]
achk1
pheno celltype approach N
1: dichPheno CD8 inbuilt 8
2: dichPheno CD8 custom 8
3: dichPheno CD8 bMIND 8
4: dichPheno CD8 swCAM 8
5: dichPheno CD8 LASSO 8
6: dichPheno CD8 RIDGE 8
7: dichPheno CD14 inbuilt 7
8: dichPheno CD14 custom 7
9: dichPheno CD14 bMIND 7
10: dichPheno CD14 swCAM 7
11: dichPheno CD14 LASSO 7
12: dichPheno CD14 RIDGE 7
13: dichPheno CD19 inbuilt 7
14: dichPheno CD19 custom 7
15: dichPheno CD19 bMIND 7
16: dichPheno CD19 swCAM 7
17: dichPheno CD19 LASSO 7
18: dichPheno CD19 RIDGE 7
19: dichPheno+sex CD4 inbuilt 9
20: dichPheno+sex CD4 custom 9
21: dichPheno+sex CD4 bMIND 9
22: dichPheno+sex CD4 swCAM 9
23: dichPheno+sex CD4 LASSO 9
24: dichPheno+sex CD4 RIDGE 9
25: dichPheno+sex CD8 inbuilt 7
26: dichPheno+sex CD8 custom 7
27: dichPheno+sex CD8 bMIND 7
28: dichPheno+sex CD8 swCAM 7
29: dichPheno+sex CD8 LASSO 7
30: dichPheno+sex CD8 RIDGE 7
31: dichPheno+sex CD14 inbuilt 7
32: dichPheno+sex CD14 custom 7
33: dichPheno+sex CD14 bMIND 7
34: dichPheno+sex CD14 swCAM 7
35: dichPheno+sex CD14 LASSO 7
36: dichPheno+sex CD14 RIDGE 7
37: dichPheno+sex CD19 inbuilt 8
38: dichPheno+sex CD19 custom 8
39: dichPheno+sex CD19 bMIND 8
40: dichPheno+sex CD19 swCAM 8
41: dichPheno+sex CD19 LASSO 8
42: dichPheno+sex CD19 RIDGE 8
43: contPheno CD8 inbuilt 9
44: contPheno CD8 custom 9
45: contPheno CD8 bMIND 9
46: contPheno CD8 swCAM 9
47: contPheno CD8 LASSO 9
48: contPheno CD8 RIDGE 9
49: contPheno CD14 inbuilt 9
50: contPheno CD14 custom 9
51: contPheno CD14 bMIND 9
52: contPheno CD14 swCAM 9
53: contPheno CD14 LASSO 9
54: contPheno CD14 RIDGE 9
55: contPheno CD19 inbuilt 9
56: contPheno CD19 custom 9
57: contPheno CD19 bMIND 9
58: contPheno CD19 swCAM 9
59: contPheno CD19 LASSO 9
60: contPheno CD19 RIDGE 9
61: contPheno+sex CD14 inbuilt 9
62: contPheno+sex CD14 custom 9
63: contPheno+sex CD14 bMIND 9
64: contPheno+sex CD14 swCAM 9
65: contPheno+sex CD14 LASSO 9
66: contPheno+sex CD14 RIDGE 9
67: contPheno+sex CD19 inbuilt 9
68: contPheno+sex CD19 custom 9
69: contPheno+sex CD19 bMIND 9
70: contPheno+sex CD19 swCAM 9
71: contPheno+sex CD19 LASSO 9
72: contPheno+sex CD19 RIDGE 9
pheno celltype approach N
ggboxplot(
data = mychktestonly[scenario == "common", ],
x = "approach", y = "auc", ylab = "AUC",
palette = appro.cols,
alpha = 0.35, fill = "approach",
add = c("jitter"),
add.params = list(shape = 21, size = 2.5, alpha = 1)
) +
scale_fill_manual(labels = appro_lab, values = appro.cols) +
facet_grid(pheno ~ celltype, scales = "free") +
labs(fill = NULL) +
used_ggthemes(
axis.text.x = element_blank(),
legend.position = "top",
strip.text.y.right = element_text(angle = 0),
legend.text = element_text(size = rel(1))
) +
rremove("xlab")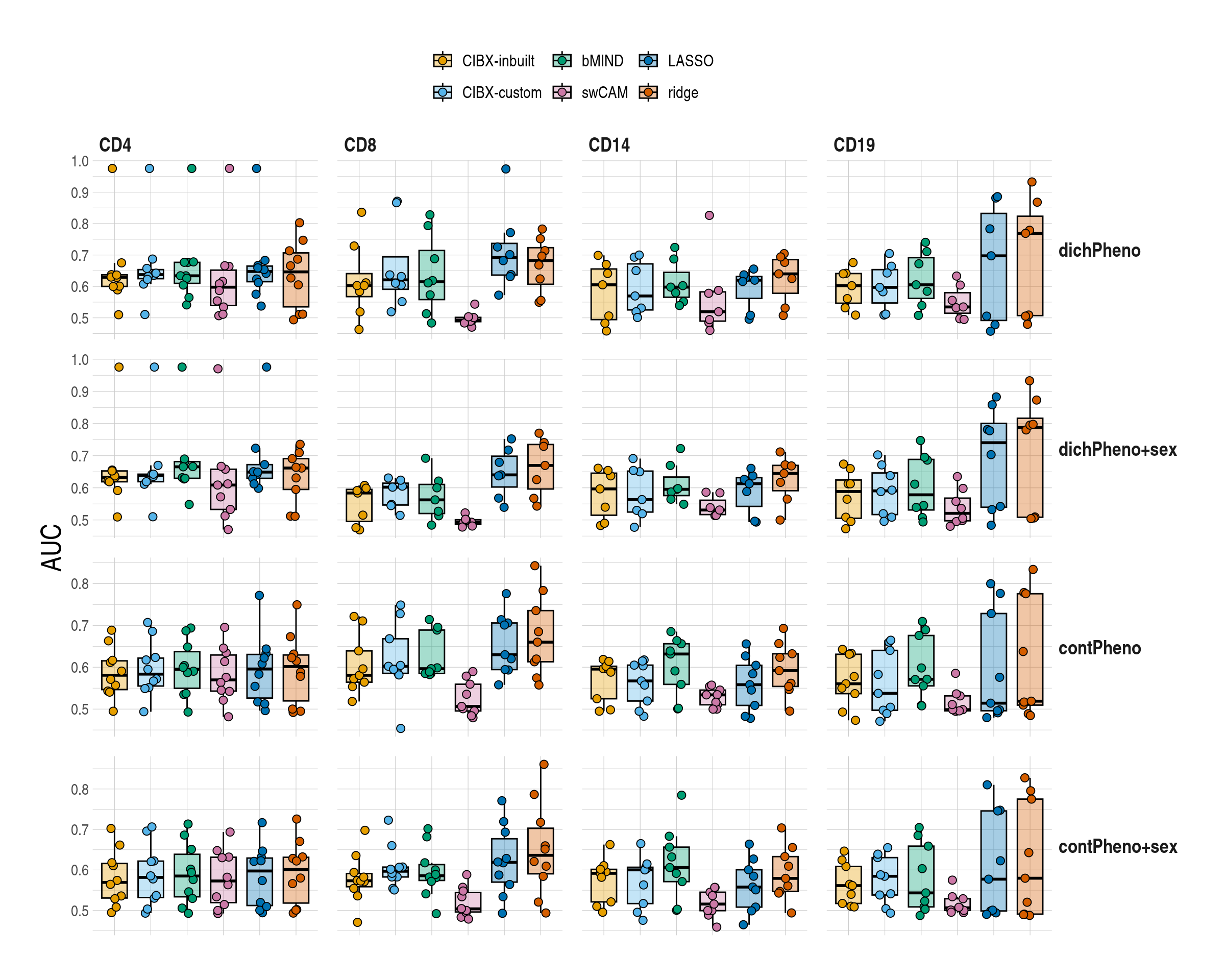
Code
tbl_out <- tbl_res[rows.order, c("celltype", grep("common", cols.order, value = TRUE))]
names(tbl_out) <- gsub("main_|common_", "", names(tbl_out))
capin <- paste0(
"Median AUC (Q25-Q75) across",
ifelse(nrow(achk1) != 0, "", uniqueN(mychktestonly$fakedPCs)),
" simulated phentoypes per cell by apparoch. **Genes common across approaches per cell type**"
)
kbl(
x = tbl_out, row.names = FALSE,
caption = capin
) %>%
kable_paper("striped", full_width = FALSE) %>%
column_spec(column = 6:7, bold = TRUE) %>%
pack_rows("dichotomous pheno", 1, 4,
label_row_css = "text-align: left;"
) %>%
pack_rows("dichotomous pheno + sex", 5, 8) %>%
pack_rows("continous pheno", 9, 12) %>%
pack_rows("continous pheno + sex", 13, 16)| celltype | inbuilt | custom | bMIND | swCAM | LASSO | RIDGE |
|---|---|---|---|---|---|---|
| dichotomous pheno | ||||||
| CD4 | 0.63 (0.6-0.64) | 0.64 (0.62-0.65) | 0.63 (0.61-0.68) | 0.6 (0.54-0.65) | 0.65 (0.62-0.66) | 0.65 (0.54-0.71) |
| CD8 | 0.6 (0.57-0.64) | 0.62 (0.59-0.69) | 0.61 (0.56-0.71) | 0.49 (0.49-0.5) | 0.69 (0.64-0.74) | 0.68 (0.61-0.72) |
| CD14 | 0.61 (0.49-0.66) | 0.57 (0.53-0.67) | 0.6 (0.57-0.64) | 0.52 (0.49-0.58) | 0.62 (0.56-0.63) | 0.64 (0.58-0.69) |
| CD19 | 0.6 (0.55-0.64) | 0.6 (0.55-0.65) | 0.61 (0.56-0.69) | 0.53 (0.51-0.58) | 0.7 (0.49-0.83) | 0.77 (0.51-0.82) |
| dichotomous pheno + sex | ||||||
| CD4 | 0.63 (0.62-0.65) | 0.64 (0.62-0.64) | 0.67 (0.63-0.68) | 0.61 (0.53-0.66) | 0.65 (0.63-0.67) | 0.66 (0.59-0.69) |
| CD8 | 0.58 (0.5-0.6) | 0.6 (0.55-0.61) | 0.56 (0.52-0.61) | 0.49 (0.49-0.5) | 0.64 (0.6-0.7) | 0.67 (0.6-0.73) |
| CD14 | 0.6 (0.51-0.65) | 0.56 (0.52-0.65) | 0.6 (0.58-0.63) | 0.53 (0.52-0.56) | 0.61 (0.54-0.63) | 0.64 (0.59-0.67) |
| CD19 | 0.59 (0.51-0.62) | 0.59 (0.52-0.65) | 0.58 (0.53-0.69) | 0.52 (0.5-0.57) | 0.74 (0.54-0.8) | 0.79 (0.51-0.82) |
| continous pheno | ||||||
| CD4 | 0.58 (0.55-0.62) | 0.58 (0.56-0.62) | 0.6 (0.55-0.64) | 0.57 (0.54-0.63) | 0.6 (0.53-0.63) | 0.6 (0.52-0.63) |
| CD8 | 0.58 (0.56-0.64) | 0.6 (0.59-0.67) | 0.6 (0.59-0.69) | 0.51 (0.5-0.56) | 0.63 (0.59-0.71) | 0.66 (0.61-0.74) |
| CD14 | 0.6 (0.52-0.6) | 0.57 (0.52-0.61) | 0.63 (0.56-0.66) | 0.53 (0.51-0.55) | 0.56 (0.51-0.6) | 0.59 (0.55-0.63) |
| CD19 | 0.56 (0.54-0.63) | 0.54 (0.5-0.64) | 0.57 (0.56-0.68) | 0.5 (0.5-0.53) | 0.51 (0.5-0.73) | 0.52 (0.51-0.78) |
| continous pheno + sex | ||||||
| CD4 | 0.57 (0.53-0.62) | 0.58 (0.53-0.62) | 0.59 (0.53-0.64) | 0.57 (0.52-0.63) | 0.6 (0.51-0.63) | 0.6 (0.52-0.63) |
| CD8 | 0.57 (0.56-0.59) | 0.6 (0.58-0.61) | 0.59 (0.57-0.61) | 0.5 (0.5-0.54) | 0.62 (0.57-0.68) | 0.64 (0.59-0.7) |
| CD14 | 0.59 (0.52-0.6) | 0.6 (0.52-0.61) | 0.61 (0.57-0.66) | 0.52 (0.5-0.55) | 0.56 (0.51-0.6) | 0.58 (0.55-0.63) |
| CD19 | 0.56 (0.52-0.61) | 0.58 (0.54-0.63) | 0.54 (0.51-0.66) | 0.51 (0.5-0.53) | 0.58 (0.5-0.75) | 0.58 (0.49-0.77) |
2.15 AUCs in pseudo datasets
testing samples + varying gene sets
Code
simres_pltlist$main$sce2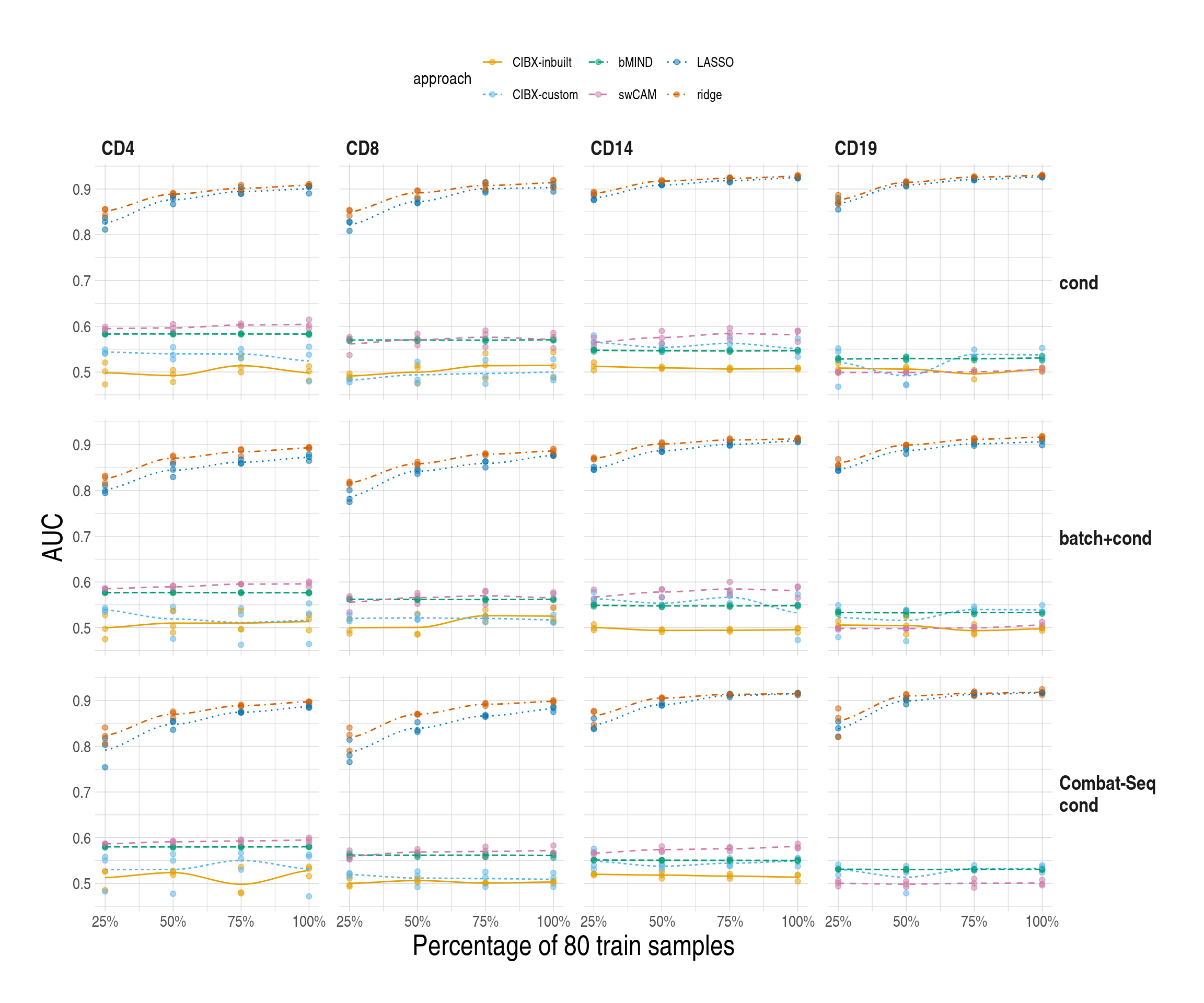
2.16 AUCs in pseudo datasets
testing samples + common gene set
Code
simres_pltlist$common$sce2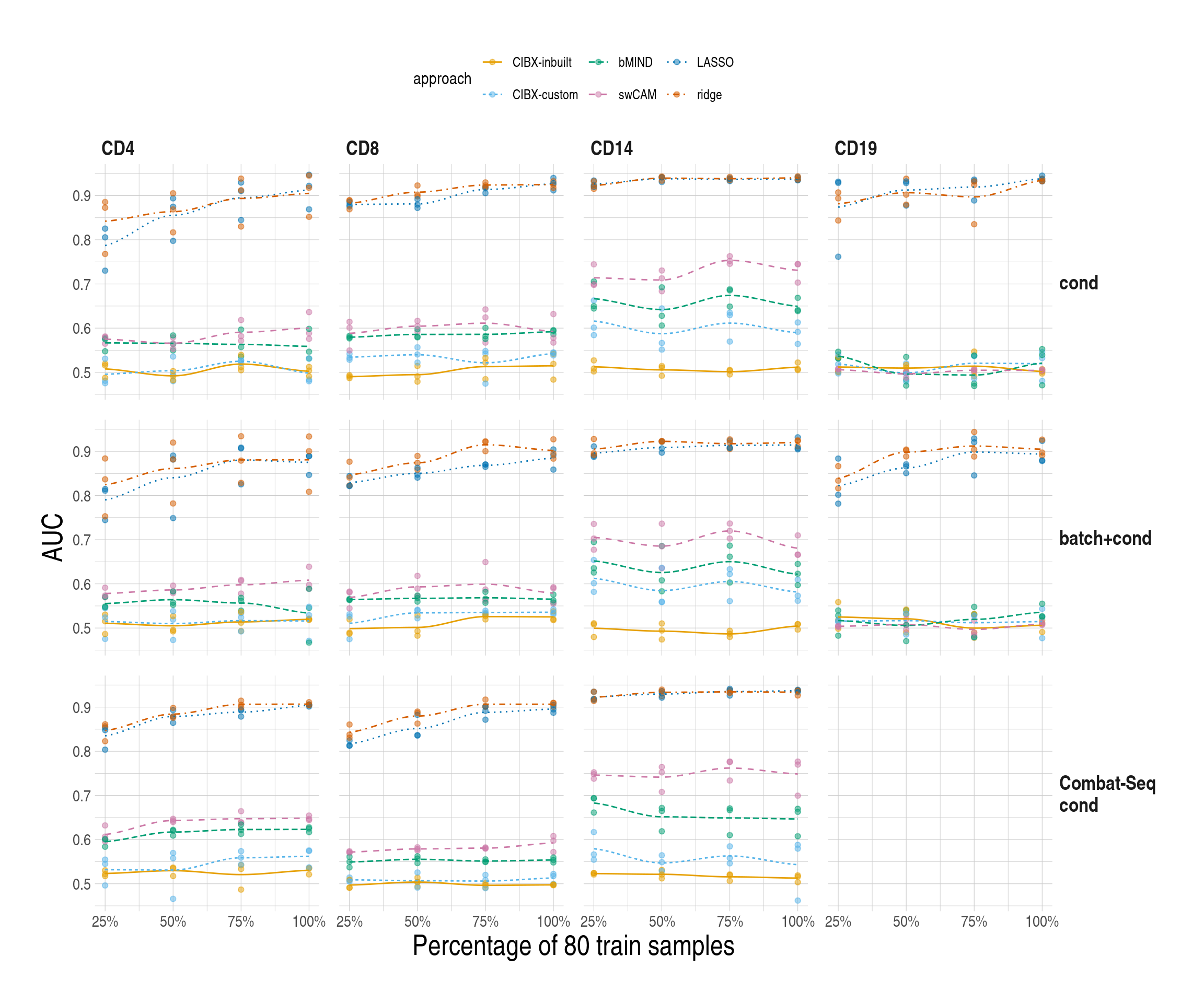
Session info
R version 4.3.3 (2024-02-29)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Rocky Linux 8.10 (Green Obsidian)
Matrix products: default
BLAS/LAPACK: /usr/lib64/libopenblas-r0.3.15.so; LAPACK version 3.9.0
locale:
[1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8
[5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8
[7] LC_PAPER=en_GB.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
time zone: GB
tzcode source: system (glibc)
attached base packages:
[1] grid parallel stats graphics grDevices datasets utils
[8] methods base
other attached packages:
[1] circlize_0.4.16 cowplot_1.1.1 pROC_1.18.4
[4] kableExtra_1.3.4.9000 cols4all_0.7-1 ggpubr_0.6.0
[7] edgeR_3.42.4 limma_3.56.2 gridExtra_2.3
[10] ggrepel_0.9.3 RColorBrewer_1.1-3 ggplot2_3.5.1
[13] ComplexHeatmap_2.18.0 styler_1.10.2 knitr_1.44
[16] stringr_1.5.0 magrittr_2.0.3 data.table_1.14.8
[19] doParallel_1.0.17 iterators_1.0.14 foreach_1.5.2
loaded via a namespace (and not attached):
[1] rstudioapi_0.15.0 jsonlite_1.8.7 shape_1.4.6
[4] farver_2.1.1 rmarkdown_2.25 GlobalOptions_0.1.2
[7] vctrs_0.6.5 rstatix_0.7.2 webshot_0.5.5
[10] htmltools_0.5.6.1 polynom_1.4-1 curl_5.1.0
[13] broom_1.0.5 htmlwidgets_1.6.2 plyr_1.8.9
[16] mime_0.12 lifecycle_1.0.4 pkgconfig_2.0.3
[19] Matrix_1.6-5 R6_2.5.1 fastmap_1.1.1
[22] shiny_1.7.5 clue_0.3-65 digest_0.6.33
[25] colorspace_2.1-0 S4Vectors_0.38.2 rprojroot_2.0.3
[28] labeling_0.4.3 spacesXYZ_1.3-0 fansi_1.0.5
[31] httr_1.4.7 abind_1.4-5 mgcv_1.9-1
[34] compiler_4.3.3 here_1.0.1 fontquiver_0.2.1
[37] withr_2.5.1 backports_1.4.1 carData_3.0-5
[40] highr_0.10 Rttf2pt1_1.3.12 R.utils_2.12.2
[43] ggsignif_0.6.4 MASS_7.3-60.0.1 rjson_0.2.21
[46] ggpp_0.5.8-1 ggsci_3.0.0 gfonts_0.2.0
[49] tools_4.3.3 zip_2.3.0 httpuv_1.6.11
[52] extrafontdb_1.0 R.oo_1.25.0 glue_1.7.0
[55] nlme_3.1-164 R.cache_0.16.0 promises_1.2.1
[58] cluster_2.1.6 generics_0.1.3 gtable_0.3.4
[61] R.methodsS3_1.8.2 tidyr_1.3.0 xml2_1.3.5
[64] car_3.1-2 utf8_1.2.3 BiocGenerics_0.46.0
[67] pillar_1.9.0 later_1.3.1 splines_4.3.3
[70] dplyr_1.1.4 lattice_0.22-5 renv_1.0.10
[73] tidyselect_1.2.0 fontLiberation_0.1.0 locfit_1.5-9.8
[76] fontBitstreamVera_0.1.1 IRanges_2.34.1 svglite_2.1.2
[79] crul_1.4.0 stats4_4.3.3 xfun_0.40
[82] matrixStats_1.0.0 pheatmap_1.0.12 stringi_1.7.12
[85] yaml_2.3.7 evaluate_0.22 codetools_0.2-19
[88] httpcode_0.3.0 extrafont_0.19 gdtools_0.3.3
[91] tibble_3.2.1 BiocManager_1.30.22 cli_3.6.2
[94] xtable_1.8-4 systemfonts_1.0.5 munsell_0.5.0
[97] Rcpp_1.0.11 png_0.1-8 ellipsis_0.3.2
[100] viridisLite_0.4.2 hrbrthemes_0.8.0 scales_1.3.0
[103] openxlsx_4.2.5.2 purrr_1.0.2 crayon_1.5.2
[106] GetoptLong_1.0.5 rlang_1.1.3 rvest_1.0.3